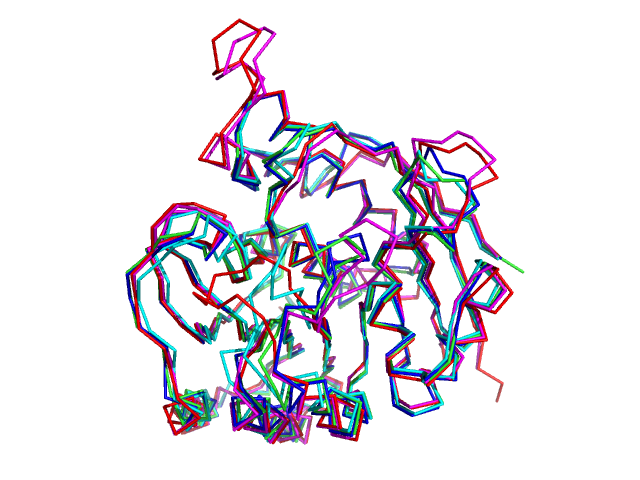Умения. d2. Пространственное выравнивание.
3. Совмещение и пространственное выравнивание.
файл для чтения PyMol
txt-файл от PDBeFold
Аналогично первому заданию этого семестра, я нашел 4 структуры, гомологичных 1vi9, с помощью PDBeFold.
Это структуры 1vi9, 3pzs, 1td2, 2ajp, 1lhp, которые представляют из себя пиридоксаль- или
пиридоксаминкиназы про- или эукариот.
Для выравнивания я взял только первую цепь, причем,
как можно видеть из рис. 1, различия между структурами в некоторых участках существенные.
- Рисунок 1. Полученное PDBeFold наложение выбранных структур.
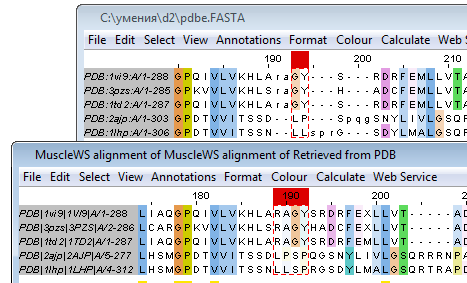
Текстовое выравнивание PDBeFold в fasta-формате я сравнил с выравниванием Muscle с параметрами
по умолчанию. Выравнивания отличаются в нескольких местах, одно из которых я предлагаю
посмотреть на рис.2 и 3.
- Рисунок 2. Множественное выравнивание по структуре (выше) и последовательности (ниже).
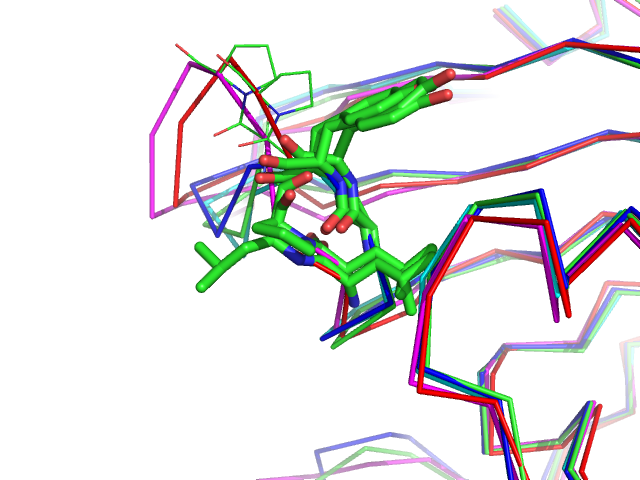
- Рисунок 3. Суперпозиция отмеченного на Рис.2 участка в структурном выравнивании. В палочном виде показаны остатки, выровненные PDBeFold, линиями представлены остатки двух последовательностей эукариот, которые Muscle выровняло с парой GY прокариот.
Как можно убедиться, структурное выравнивание дает более правильный результат
(тот же вывод я сделал и по другим подобным сайтам).
4. совмещение pair_fit в PyMol.
Альфа и бета-цепочки Т-клеточного рецептора были взяты из структуры 1mi5. Файлы:
region d:118-206 - альфа-цепочка.
region e:119-247 - бета-цепочка.
Для выполнения задания с сервером SheeP понадобилась команда аналогичная
save betha.pdb, betha
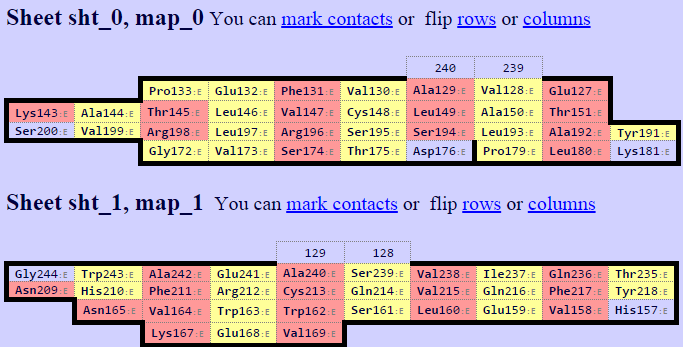
На сервере SheeP были построены карты листов для альфа-цепочки (Рис.4) и бета-цепочки (Рис.5).
Ход тяжей однозначно задает соответствие между листом альфа-цепи и первым листом бета-цепи.
Для получения одинакового расположения я один раз использовал “flip columns”.
- Рисунок 4. Карта бета-листа альфа-цепочки.
- Рисунок 5. Карты двух бета-листов в бета-цепочке.
По рисункам 4 и 5 удобно определить множества остатков, по которым можно
совместить две структуры. Это делает следующая команда в PyMol:
pair_fit (alpha and /////CA and (i. 127-130 or i. 136-139 or i. 158-160 or i. 142+177)), (betha and /////CA and (i. 130-133 or i. 145-148 or i. 173-175 or i. 151+192))
или немного короче
pair_fit alpha ///127-130+136-139+158-160+142+177/CA, betha///130-133+145-148+173-175+151+192/CA
Я старался охватить все края бета-листа, чтобы получить наложение с минимальным RMS по остаткам,
надеясь, что остатки между выбранными участками тоже лягут хорошо (насчет некоторых из них не было
полной уверенности, так как изображение SheeP показывало, что там можно предполагать выпетливания
в некоторых тяжах, см. рис.4,5).
В итоге PyMol выдал RMS = 1.394 (13 to 13 atoms), что хорошо.
Приятно было
увидеть, что и остальные части листа, а главное петли между его тяжами и даже некоторые нерегулярные
цепи имели хорошее наложение (Рис. 6, 7 и 8).
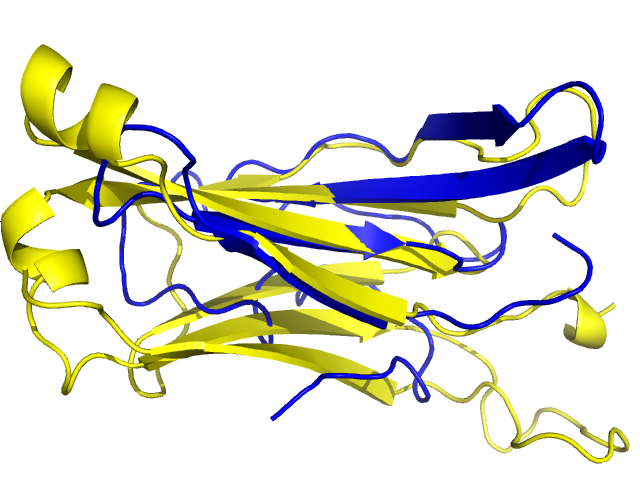
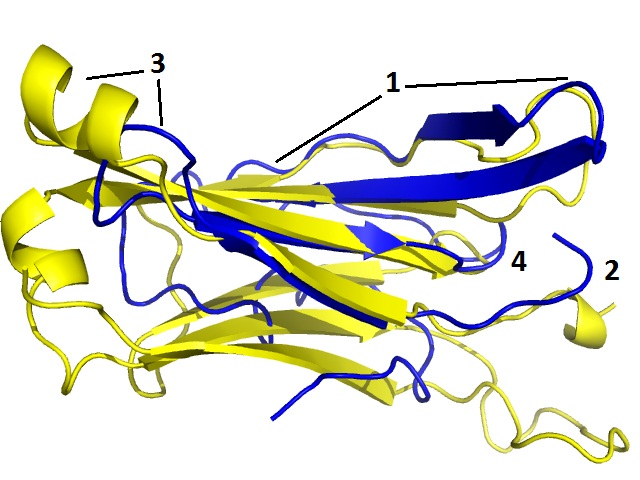
Синим либо голубым (выравниваемые атомы, рис. 8) на рисунках 6-8 показана альфа-цепь,
бета-цепь показана желтым или оранжевым (выравниваемые атомы).
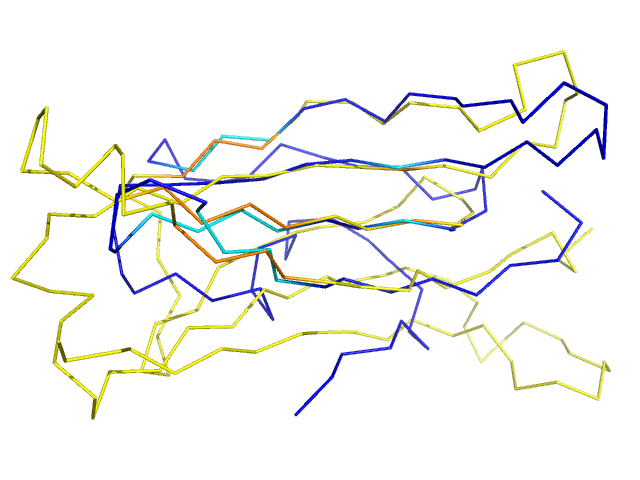
На рисунке 9 показаны отдельные элементы структуры цепей, которые не выравнивались,
но тем не менее хорошо совместились. По ним можно еще раз убедиться, как хорошо совпадает
ход цепей белка, что было предсказано по результатам программы SheeP (Рис 4, 5).
- Рисунок 6. наложение pair-fit выбранных цепей Т-клеточного рецептора в виде "cartoon".
- Рисунок 7. Участки структур, которые не принимали участия в выравнивании, но при этом хорошо соответствуют друг другу.
- Рисунок 8. наложение pair-fit альфа- и бета-цепей Т-клеточного рецептора, указаны атомы, по которым строилось совмещение.
Вы можете лично убедиться в качестве выравнивания двух листов, а также соседних участков цепи, в PyMol: сессия PyMol.
На страницу 7 семестра
© Aleshin Vasily