Сравнение матриц аминокислотных замен
Матрица весов аминокислотных замен – это симметричная таблица, в которой по вертикали и по горизонтали в одинаковом порядке расположены названия аминокислот. В местах пересечения столбцов и строчек, соответствующих определенным аминокислотам располагаются числа – веса аминокислотных замен. Они дают относительную оценку заменам аминокислот, исходя из частот встречаемости этих замен в природе.
Матрицы типа BLOSUM построены с использованием базы данных BLOKS, в ней содержатся множественные сопоставления отдельных участков (блоков) белков из различных семейств. При построении матриц учитывается частота встречаемости различных замен аминокислот, а также и то, что эти частоты могут значительно сдвигаться из-за наличия почти идентичных последовательностей в BLOKS. Для того, чтобы избежать сдвига, похожие последовательности кластеризуют (группируют в кластер), а веса замен в кластере считают относительно его величины. Для определения «схожести» последовательностей вводят число – порог идентичности. Если процент идентичности выравнивания двух последовательностей больше, чем порог идентичности, то они группируются в кластер. Причем для попадания в кластер последовательности нужно преодолеть этот порог в сравнении хотя бы с одной последовательностью из кластера. Таким образом, в кластере могут оказаться и последовательности со значением идентичности много меньше, чем порог. Не для любых белков можно применять матрицу BLOSUM, так как в базе данных BLOKS собраны в основном последовательности цитоплазматических белков. Если белок локализован в мембране, то веса замен аминокислот могут существенно отличаться. Поэтому для таких белков созданы специальные матрицы, например матрица PHAT. В этих матрицах частота встречаемости замен описывается, основываясь на выравниваниях трансмембранных участков белков. В ходе выполнения задания матрица BLOSUM62 была реконструирована с использованием последней весрии базы данных BLOKS. Далее эта матрица будет обозначаться BLOSUM62*.
Матрица BLOSUM62*
Таблица 1. Сравнение весов замен для изолейцина в различных матрицах
| Трехбуквенное обозначение аминокислоты | Однобуквенное обозначение аминокислоты | Вес по матрице BLOSUM62 | Вес по матрице PHAT | Вес по матрице BLOSUM62* |
| Ile | I | 5 | 4 | 4 |
| Leu | L | 2 | 2 | 2 |
| Val | V | 3 | 3 | 2 |
| Ala | A | -1 | 0 | -2 |
| Gly | G | -4 | -2 | -4 |
| Arg | R | -3 | -6 | -3 |
| His | H | -3 | -5 | -3 |
Замена аминокислоты Ile на саму себя
Величины замены аминокислоты Ile на саму себя для матриц BLOSUM62 – 4, а для матрицы PHAT – 5. Эти значения отличаются, так как матрицы BLOSUM применяются для цитоплазматических белков, а PHAT для трансмембранных участков. Мембранные белки должны обладать достаточно консервативным расположением гидрофобных аминокислот в своей структуре для заякоривания в мембране. Поэтому гидрофобные аминокислоты в мембранных белках изменяться будут реже, а встречаться друг против друга в выравниваниях чаще. Это приводит к увеличению значения веса их замен на самих себя в матрице PHAT.
Замена аминокислоты Ile на близкие по химическим свойствам
Изолейцин - алифатическая гидрофобная аминокислота. Также к этой группе относятся лейцин, валин и аланин. В таблице 1 приведены значения весов замен Ile на эти аминокислоты. Как видно из таблицы, вес по матрице PHAT всегда больше или равен весам по матрицам BLOSUM, а веса по матрицам BLOSUM одинаковые (за исключением Ala, возможно это связано со статистическими факторами, так как в матрице BLOSUM62* использована более новая версия BLOKS). Эти результаты можно объяснить так же, как и в случае замены аминокислоты Ile на саму себя. Замена гидрофобной аминокислоты на гидрофобную в мембранном белке будет встречаться немного чаще, чем в цитоплазматическом.
Замена аминокислоты Ile на аминокислоты из других функциональных групп
Как видно из таблицы 1, замена изолейцина на глицин в мембранных белках сохраняется в большем числе случаев, чем в цитоплазматических. Возможно, это связано с тем, что глицин не является заряженной аминокислотой и может беспрепятственно находится в мембране. В случае трансмемранных белков - это преимущество. Однако для цитоплазматических белков такого преимущества у глицина нет, и его сохранение менее благоприятно. Необходимо отметить, что во всех трех случаях подобная замена сохраняется дочтаточно редко - все веса замен отрицательные.
Для аргинина и гистидина наблюдается обратная картина. Эти аминокислоты положительно заряженные, и частота замены изолейцина на них в мембранных белках меньше, чем в цитоплазматических. Положительно заряженная аминокислота будет препятствовать нахождению белка в мембране, а значит, он перестанет выполнять свою функцию. Все веса замен изолейцина на гистидин и аргинин большие по модулю и отрицательные, это означает, что такие замены встречаются крайне редко.
Сравнение выравниваний, полученных для коротких мутантов вручную и построенных классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана
Краткое описание программ needle и water пакета EMBOSS
Программа needle реализует алгоритм Нидлмана-Вунша, предназначенный для глобального парного выравнивания. Глобальное выравнивание означает, что последовательности сравниваются друг с другом целиком, и если одна последовательность меньше другой, то она будет дополнена до размера большей с помощью gap. Программа water реализует алгоритм Смита-Ватермана, который выполняет локальное парное выравнивание. В случае локального выравнивания программа находит оптимальное по весу выравнивание на определенном участке последовательности. Вне этого участка, в отличие от программы needle, пробелы (gap) не учитываются. Таким образом, при выравнивании короткой и длинной последовательностей процент сходства в программе water будет больше. После выполнения алгоритма обе программы записывают в файл результат с самым оптимальным весом выравнивания.
Подробнее об этих программах можно узнать, пройдя по ссылкам:
EMBOSS: water
EMBOSS: needle
Основные параметры, используемые программами needle и water
Для параметров в квадратных скобках значения задаются без указания названия параметра, просто записываются по порядку или при запросе программы.
| Параметр | Краткое описание |
| [-asequence] и [-bsequence] |
USA сравниваемых последовательностей |
| [-outfile] | Имя файла с выравниванием, который программа создает после выполнения алгоритма |
| -gapopen | Штраф за открывающий gap. Значение по умолчанию 10.0 |
| -gapextend | Штраф за каждый gap, следующий за открывающим. Значение по умолчанию 0.5 |
| -datafile | Используемая матрица весов замен. По умолчанию BLOSUM62 для белков и DNAFULL для ДНК |
В связи с тем, что программа needle выполняет глобальное выравнивание, для неё важны также следующие параметры:
| -endweight | Штраф за gap на конце выравнивания. По умолчанию отключена |
| -endopen | Штраф за открывающий gap на конце выравнивания. По умолчанию 10.0 |
| -endextend | Штраф за каждый gap, следующий за открывающим на конце. Значение по умолчанию 0.5 |
Сравнение выравниваний, полученных для коротких мутантов вручную и построенных классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана
С помощью скрипта evolve_protein.pl были созданы 3 последовательности mut1, mut2, mut3, с вероятностью изменения остатка (модель "ошибки" ДНК-полимеразы) 0,6, 0,6 и 0,4 соответственно, и с вероятностью замены остатка в случае изменения данной позиции 0,6, 0,8 и 0,8 соответственно. Таким образом, в последовательности mut1 вероятность мутации такая же, как и в mut2 и выше, чем в mut3, однако в mut1 вероятность возникновения инсерции или замены ниже, чем в mut2. В mut3 вероятность мутации ниже, чем в mut1 и в mut2, а также там с меньшей вероятностью произойдет инсерция или замена. В JalView мной была настроена схема цветов отображения аминокислот. Согласно этой схеме гидрофобные аминокислоты Gly, Ala, Val, Leu, Ile покрашены оранжевым, серосодержащие аминокислоты Met, Cys – желтым, гидрофобные Phe, Tyr, Trp – серым, нейтральные Ser и Thr – розовым, нейтральные Asn, Gln – темно-зеленым, иминокислота Pro – белым, кислые Asp, Glu – светло-зеленым, основные His, Lys, Arg – фиолетовым. Далее 3 последовательности mut1, mut2, mut3 в формате fasta были по очереди вручную выровнены с исходной последовательностью.
Сравнение выравниваний, полученных с помощью ручного выравнивания и программ water и needle для mut1
Таблица 1. Сравнение выравниваний для mut1.
| - | Изображение участка выравнивания | identity | similarity | Вес | Положение выравнивания относительно длинной последовательности |
| Выравнивание, сделанное вручную | 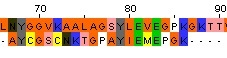 |
30% | 40% | 19.0 | 67-86 |
| Выравнивание, полученное с помощью программы water |  |
44.4% | 77.8% | 22.0 | 78-86 |
| Выравнивание, полученное с помощью программы needle | 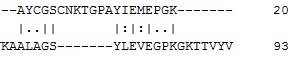 |
2.9% | 3.8% | 21.0 | 74-86 |
На таблице 1 показано сравнение выравниваний, полученных с помощью ручного выравнивания и программ water и needle для mut1. Участок выравнивания с 79ой по 86ю аминокислоты совпадает во всех трех случаях. Вес выравнивания различается незначительно, однако identity и similarity различны. Выравнивание, полученное с помощью программы water имеет значения identity и similarity большие, чем выравнивание, полученное вручную. Это объясняется тем, что программа water выполняет локальное выравниваение. В данном случае оно построено не для кусочка в 20 аминокислот, как для ручного выравнивания, а для кусочка в 8 аминокислот, и так как значения identity и similarity вычиляются относительно размера последовательности, они отличаются для выравниваний разной длины. Значения identity и similarity для выравнивания, полученного с помощью программы needle значительно меньше, потому что программа needle осуществляет глобальное выравнивание, и эти значения вычисляются относительно полной длины последовательности.
Сравнение выравниваний, полученных с помощью ручного выравнивания и программ water и needle для mut2
Таблица 2. Сравнение выравниваний для mut2.
| - | Изображение участка выравнивания | identity | similarity | Вес | Положение выравнивания относительно длинной последовательности |
| Выравнивание, сделанное вручную | 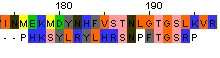 |
25% | 30% | 9.0 | 176-195 |
| Выравнивание, полученное с помощью программы water | 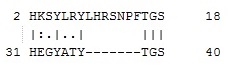 |
35.3% | 41.2% | 23.0 | 31-40 |
| Выравнивание, полученное с помощью программы needle | 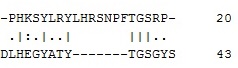 |
2.5% | 2.9% | 15.0 | 30-42 |
На таблице 2 показано сравнение выравниваний, полученных с помощью ручного выравнивания и программ water и needle для mut2. Для значений identity и similarity наблюдается такая же картина, как и в случае mut1. Однако участки выравнивания не совпадают. Можно сделать вывод, что ручное выравнивание было сделано неправильно, так как участки выравниваний, полученных программами needle и water совпадают, и их вес заметно больше, чем в случае ручного выравнивания.
Сравнение выравниваний, полученных с помощью ручного выравнивания и программ water и needle для mut3
Таблица 3. Сравнение выравниваний для mut3.
| - | Изображение участка выравнивания | identity | similarity | Вес | Положение выравнивания относительно длинной последовательности |
| Выравнивание, сделанное вручную | 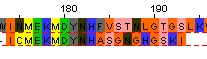 |
50% | 55% | 39.0 | 174-193 |
| Выравнивание, полученное с помощью программы water |  |
60% | 70% | 50.5 | 174-193 |
| Выравнивание, полученное с помощью программы needle |  |
5.6% | 6.9% | 48.5 | 174-196 |
На таблице 3 показано сравнение выравниваний, полученных с помощью ручного выравнивания и программ water и needle для mut3. Для значений identity и similarity наблюдается такая же картина, как и в случаях mut1 и mut2, но в этом случае участки выравнивания совпадают. Различия веса и значений identity и similarity для ручного выравнивания и полученного с помощью water различаются из-за того, что программа water смоделировала делецию 185й и 186й аминокислот исходного пептида, и не стала включать в выравнивание последние две аминокислоты mut3. В этом случае постановка gap оказалась целесообразной, что было не очевидно в ручном выравнивании. Значения identity и similarity для выравнивания посредством needle ниже, чем в двух других случаях. Это согласуется с результатами для mut1 и mut2. Зато вес в третьем случае лишь ненамного ниже, чем во втором. Небольшое уменьшение наблюдается из-за постановки gap, которая не до конца компенсируется весом совпадения лизина 195. Такое различие в значениях identity, similarity и в весе выравниваний, полученных с помощью программ needle и water наглядно иллюстрирует различия локального и глобального выравниваний.