Ознакомление с программой Muscle
Получил файл с последовательностями 34 дельта-антигенов в формате fasta delta.fasta с помощью SRS (для чего при составлении запроса к банку данных Swiss-Prot ввел в поле поиска Description слово "delta", а в поле Taxonomy название рода "Deltavirus" (полный запрос выглядит так: ([swissprot-Taxonomy:Deltavirus*] & [swissprot-Description:delta*])), после чего сохранил последовательности антигенов с помощью кнопки Save).Импортировал файл в Genedoc. Так выглядит "выравнивание" невыравненных последовательностей: delta.msf:

C помощью программы Muscle построил множественное выравнивание последовательностей: delta_aligned.fasta.
Импортировал файл в Genedoc: delta_aligned.msf:

Как видно, последовательности настолько похожи между собой, что даже если просто поставить последовательности друг под другом (как в файле delta.msf), в них найдутся несколько столбцов с консервативными остатками (в начале выравнивания), сохранившими свои позиции во всех дельта-антигенах. Естественно, после выравнивания таких столбцов оказалось гораздо больше (в файле delta_aligned.msf). По результатам выравнивания можно с уверенностью сказать, что белки гомологичны и очень схожи по пространственной структуре. Такое огромное количество консервативных остатков - достаточное основание так полагать. Очевидно, в этих белках не имеют особенного биологического смысла лишь концы некоторых последовательностей (начиная с 197-й позиции выравнивания), ведь в других белках им соответсвуют гэпы (впрочем, концы белков, у которых они есть, очень сходны между собой, так что, возможно, тоже выполняют какую-то биологическую функцию, присущую лишь этим белкам).
Выравнивание набора гомологов белка DPS_ECOLI
Посредством BLAST получил 8 гомологов моего белка, встречающихся в таксоне Bacteria, E-value которых менее 0.001, а процент идентичности выравнивания менее 80%, но более 30%.Среди этих гомологов оказались:
DPS_YERPS (Q669E1) с E-value, равным 1.10-75, и процентом идентичности, равным 79%. Таксономия бактерии, содержащей этой белок: Bacteria; Proteobacteria; Gammaproteobacteria; Enterobacteriales; Enterobacteriaceae; Yersinia.
DPS_SERMA (Q84AP0) с E-value, равным 9.10-74, и процентом идентичности, равным 76%. Таксономия бактерии, содержащей этой белок: Bacteria; Proteobacteria; Gammaproteobacteria; Enterobacteriales; Enterobacteriaceae; Serratia.
DPS_SODGM (Q2NU13) с E-value, равным 6.10-72, и процентом идентичности, равным 76%. Таксономия бактерии, содержащей этой белок: Bacteria; Proteobacteria; Gammaproteobacteria; Enterobacteriales; Enterobacteriaceae; Sodalis.
DPS_ERWCT (Q6D3H7) с E-value, равным 2.10-71, и процентом идентичности, равным 74%. Таксономия бактерии, содержащей этой белок: Bacteria; Proteobacteria; Gammaproteobacteria; Enterobacteriales; Enterobacteriaceae; Pectobacterium.
DPS_ACIAD (Q6FCX7) с E-value, равным 1.10-51, и процентом идентичности, равным 60%. Таксономия бактерии, содержащей этой белок: Bacteria; Proteobacteria; Gammaproteobacteria; Pseudomonadales; Moraxellaceae; Acinetobacter.
DPS_AGRT5 (Q8UCK6) с E-value, равным 4.10-45, и процентом идентичности, равным 55%. Таксономия бактерии, содержащей этой белок: Bacteria; Proteobacteria; Alphaproteobacteria; Rhizobiales; Rhizobiaceae; Rhizobium/Agrobacterium group; Agrobacterium.
DPS_BREBE (P83695) с E-value, равным 6.10-12, и процентом идентичности, равным 33%. Таксономия бактерии, содержащей этой белок: Bacteria; Firmicutes; Bacillales; Paenibacillaceae; Brevibacillus.
DPS_STRMU (Q9KWH3) с E-value, равным 6.10-11, и процентом идентичности, равным 27%. Таксономия бактерии, содержащей этой белок: Bacteria; Firmicutes; Lactobacillales; Streptococcaceae; Streptococcus.
С помощью программы seqret пакета EMBOSS получил последовательности белка DPS_ECOLI и найденных гомологов в fasta-формате: myproteins11.fasta.
После этого с помощью программы muscle получил множественное выравнивание белка DPS_ECOLI и гомологов: myproteins_aligned11.fasta.
Затем импортировал выравнивание в GeneDoc и сохранил в файле myproteins_aligned11.msf:
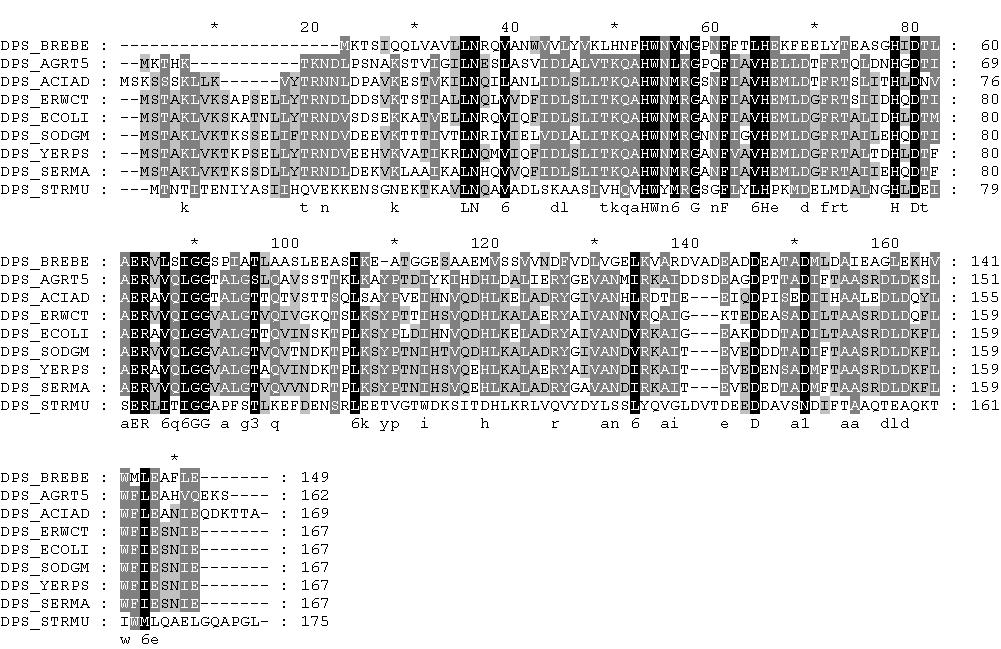
Как видно, последовательности очень сходны между собой, что говорит об очень высокой вероятности того, что они гомологичны. В выравнивании можно обнаружить очень много консервативных остатков. Самые крупные участки, состоящие почти исключительно из консервативных остатков - это, во-первых, участок с 43-й по 58-ю позицию выравнивания (сооветствует участку с 41-й по 56-ю позицию последовательности белка DPS_ECOLI), во-вторых, участок с 61-й по 75-ю позицию выравнивания (соответствует участку с 59-й по 73-ю позицию последовательности белка DPS_ECOLI), в-третьих, участок с 83-й по 98-ю позицию выравнивания (соответствует участку с 81-й по 96-ю позицию последовательности белка DPS_ECOLI), ну и, наконец, участок со 149-й по 172-ю позицию выравнивания (соответствует участку со 144-й по 167-ю позицию последовательности белка DPS_ECOLI). Безусловно, такое огромное количество больших участков выравнивания с консервативными остатками говорит о гомологичности последовательностей. Более того, по данным UniProt в последовательности белка DPS_ECOLI три аминокислотных остатка отвечают за связывание полипептидной цепи с ионом железа, что чрезвычайно важно для выполнения функции белков семейства DPS. Это аминокислотные остатки под номерами 51 (H), 78 (D) и 82 (E). Как видно из выравнивания, эти остатки консервативны для всех выравниваемых белков (они соответствуют 53-й, 80-й и 84-й позициям выравнивания), что так же говорит об их гомологичности, ведь это уже дает основания подозревать белки в выполнении схожих функций (и образовании схожих активных центров).
Однако, если присмотреться к этому выравниванию, то можно обнаружить участки с недостоверным выравниванием, скорее всего лишенные биологического смысла. Прежде всего стоит обратить внимание на начало последовательностей. Довольно очевидно, что начало последовательности белка DPS_ACIAD выравнено неверно, немного сдвинуто относительно всех остальных последовательностей. Кроме того, обращает на себя внимание участок выравнивания с 8-й по 18-ю позицию (в белке DPS_ECOLI это участок с 6-й по 16-ю позицию), где в последовательности белка DPS_AGRT5 стоят гэпы (и почти на тех же позициях гэпы стоят и в последовательности белка DPS_ACIAD). Конечно, возможно, что этот участок у других белков играет какую-либо роль в выполнении их функций, а белки DPS_AGRT5 и DPS_ACIAD, соответственно, этой функции лишены, но в это верится довольно слабо (тем более у других белков этот участок почти не несет консервативных на 100% остатков). Та же ситуация наблюдается на участке с 143-й по 145-ю позицию выравнивания (между 140-й и 141-й позициями последовательности белка DPS_ECOLI), только здесь, наоборот, гэпы стоят в последовательностях всех белков, кроме DPS_AGRT5. Ну и, наконец, внимание обращает на себя конец выравнивания. Начиная с 173-й позиции выравнивания (после 167-й позиции последовательности белка DPS_ECOLI) в последовательностях 5 белков из 7 стоят гэпы, но в посдеовательностях белков DPS_AGRT5 и DPS_ACIAD имеются аминокислотные остатки, причем не схожие между собой. Эти участки, скорее всего, не имеют никакого биологического смысла, и, вероятно, даже не влияют на пространственную структуру белков, никак не мешая выполнению их функций.