Оптимизация структуры порфирина с помощью программы Mopac
Создадим файл 1.smi с аннотацией порфирина в виде SMILES и названием молекулы (porphyrin) через несколько пробелов. Подадим его на вход программе obgen для построения 3D структуры порфирина:obgen 1.smi > 1.molВ результате получаем файл 1.mol. Просмотрим полученную структуру в PyMol и удалим ненужные водороды, сохраним результат в файле por.pdb. Изображение молекулы порфирина представлено ниже:
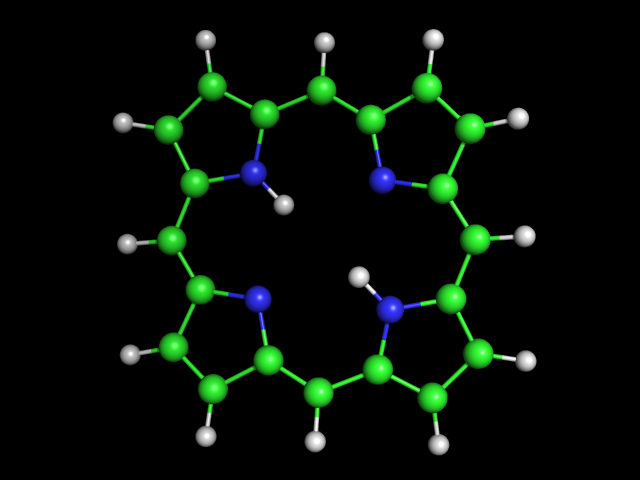
Сбоку молекула порфирина выглядит так:
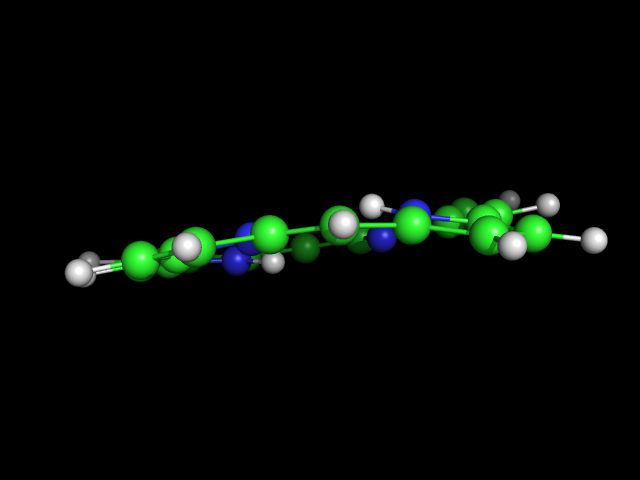
Как видно, построенная молекула не является плоской (хотя должна быть такой). C помощью babel переформатируем координаты в mol формате во входной файл для Mopac (зададим параметризацию типа pm6):
babel -ipdb por.pdb -omop 1_opt.mop -xk "PM6"На выходе получаем файл 1_opt.mop. Теперь запустим Mopac:
MOPAC2009.exe 1_opt.mopЧтобы просмотреть результат оптимизации в PyMol, переформатируем файл 1_opt.out в pdb:
babel -imopout 1_opt.out -opdb 1_opt.pdbВ результате получаем файл 1_opt.pdb. Посмотрим на оптимизированную структуру молекулы порфирина сбоку:

Как видно, теперь молекула является плоской.
Попробуем провести оптимизацию с другой параметризацией (AM1):
babel -ipdb por.pdb -omop 1_opt2.mop -xk "AM1"Как и в предыдущий раз, запустим Mopac и переформатируем полученный файл в pdb. В итоге получаем файл 1_opt2.pdb.
Посмотрим на оптимизированную структуру молекулы порфирина сбоку:

Как видно, молекула, оптимизированная с параметризацией AM1, тоже является плоской. Посмотрим теперь на структуры, оптимизированные с двумя разными параметризациями, вместе:
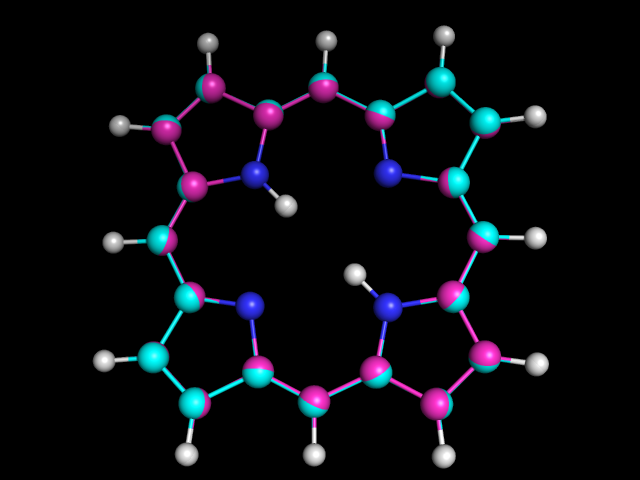
Стуктуры накладываются практически идеально.
Расчет возбужденных состояний порфирина и спектра поглощения молекулы
Для расчёта возбуждённых состояний сделаем копию mop-файла из предыдущего занятия и назовем его 1_opt_spectr.mop. Для указания Mopac о необходимости расчёта возбуждённого состояния добавим в конец файла пустую строку и строку:cis c.i.=4 meci oldgeoЗапустим MOPAC:
MOPAC2009.exe 1_opt_spectr.mopРассмотрим выходной файл 1_opt_spectr.out и найдем в нем значения энергий для электронных переходов:
STATE ENERGY (EV) Q.N. SPIN SYMMETRY POLARIZATION
ABSOLUTE RELATIVE X Y Z
1+ 0.000000 0.000000 1+ SINGLET ????
2 1.913312 1.913312 1 TRIPLET ????
3 2.266014 2.266014 2 SINGLET ????
4 2.463186 2.463186 2 TRIPLET ????
5 2.823915 2.823915 3 TRIPLET ????
6 3.362161 3.362161 4 TRIPLET ????
7 3.389757 3.389757 3 SINGLET ???? 0.2031 0.2347 0.0010
8 3.669242 3.669242 4 SINGLET ???? 2.3899 2.0438 0.0085
9 3.871323 3.871323 5 SINGLET ???? 1.5461 1.7992 0.0084
На основании этих значений рассчитаем длины волн, при которых происходят эти переходы:
| Энергия (эВ) | Длина волны (нм) |
| 1,913312 | 648,913002 |
| 2,266014 | 547,910575 |
| 2,463186 | 504,0516769 |
| 2,823915 | 439,6637412 |
| 3,362161 | 369,2782808 |
| 3,389757 | 366,2719876 |
| 3,669242 | 338,3731664 |
| 3,871323 | 320,7102672 |
Определение геометрии молекулы пара-бензохинона
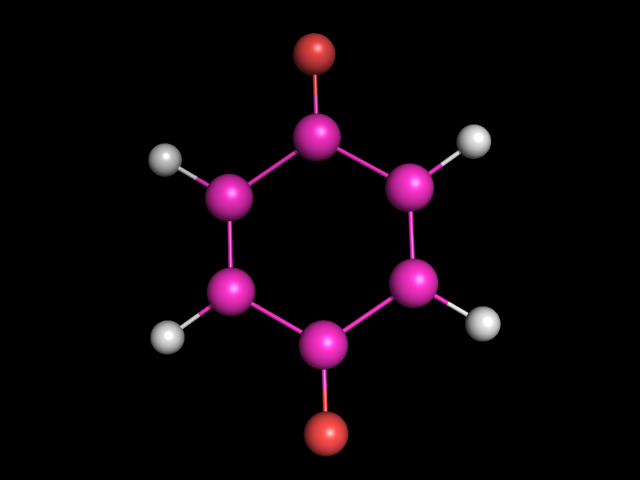
Определим геометрию молекулы O=C1C=CC(=O)C=C1 (пара-бензохинон) как с помощью obgen, так и с помощью Мopac (для Mopac будем ипользовать параметризацию pm6). На выходе получаем файлы hin.pdb (результат программы obgen) и 2_opt.pdb (результат программы Mopac). Изображение пара-бензохинона, полученное с помощью obgen, представлено ниже: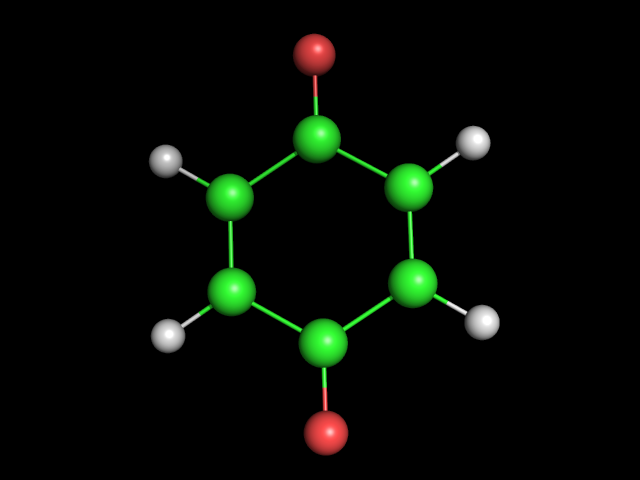
С помощью Mopac2009 получено следующее изображение:
Наложим одну структуру на другую:
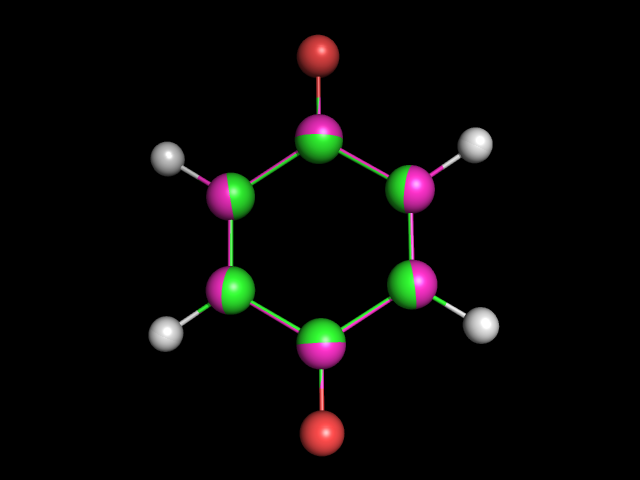
Видно, что структуры различаются незначительно.
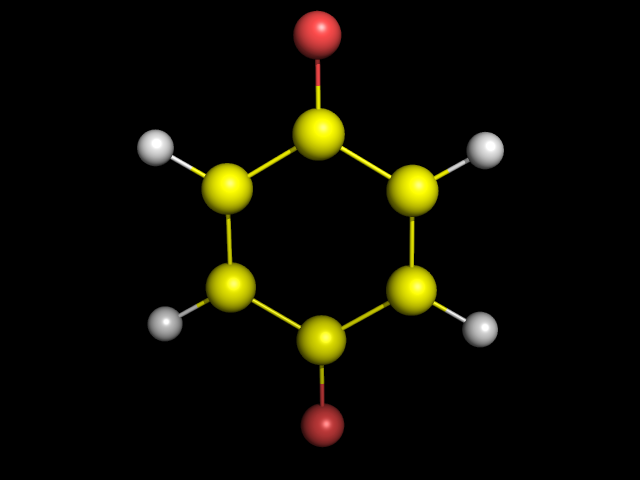
Определим теперь геометрию дианиона этой молекулы. Для этого добавим в первую строчку mop-файла слово CHARGE=-2 и явным способом укажем, на каких атомах должен находиться отрицательный заряд (допишем после символов O "(-)"). Сохраним изменения в файле 2_opt2.mop.
Запустим Mopac. В результате переформатирования получаем файл 2_opt2.pdb. Рассмотрим структуру дианиона в PyMol:
Сравним ее со структурой незаряженной молекулы, полученной с помощью Mopac:
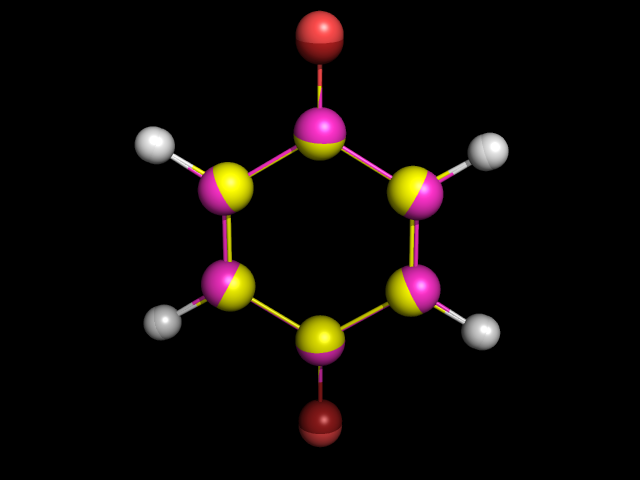
Из наложения видно, что в молекуле дианиона связи С-O более вытянуты (из-за отталкивания отрицательно заряженных атомов кислорода) - на рисунке атомы кислорода дианиона светлее, чем атомы кислорода незаряженной молекулы. Кроме того, молекула дианиона немного уже, чем незаряженная молекула (С-С связи молекулы дианиона окрашены желтым, С-С связи незаряженной молекулы - фиолетовым).
Построение структуры связывания АТФ с белком через координацию иона магния
Дана конформация, где АТФ связывается с белком через координацию иона магния, но магния в самой структуре нет. Структура сохранена в файле test.pdb.Для начала добавим в структуру атомы водородов. Но для фосфатной группы важен pH среды. Поэтому выполним эту операцию с помощью программы babel:
babel -ipdb test.pdb -opdb test2.pdb -p 7.4На выходе получаем файл test2.pdb с добавленными атомами водородами при pH, равном 7.4 (оптимальный pH для клетки).
Теперь нужно добавить в файл атом магния. Для этого скопируем атом фосфора, поменяем имя атома на Mg и запишем его координаты, как среднее арифметическое между координатами γ-фосфора и Cα аспартата. Сохраним изменения в файле test3.pdb.
Переведем полученные pdb координаты в форматы mop и xyz (с помощью babel). Получаем на выходе файлы test_opt.mop и test_opt.xyz.
Затем укажем запрет на движение для всех атомов, кроме γ-фосфата, воды и магния (чтобы потом легко восставновить конформацию в белке). Для этого поменяем в mop-файле "1" после координаты на "0". Чтобы определить атомы, которым нужно разрешить двигаться, откроем xyz файл в PyMol и отобразим labels как atoms id. Номер строчки с этим атомом в mop файле будет id+3. Сохраним изменения в файле test_opt2.mop.
Наконец, воспользуемся программой Mopac (оптимизируем с параметризацией PM6). В результате переформатирования с помощью babel получаем файл test_opt.pdb с оптимизированной структурой.
Изображение структуры представлено ниже:
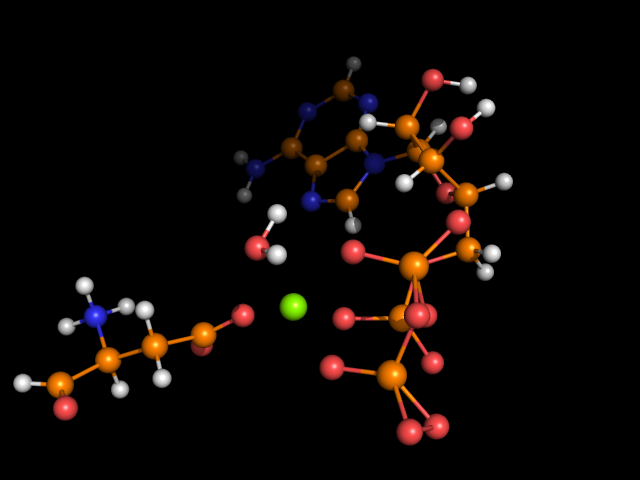
Наложим оптимизированную структуру на исходную:
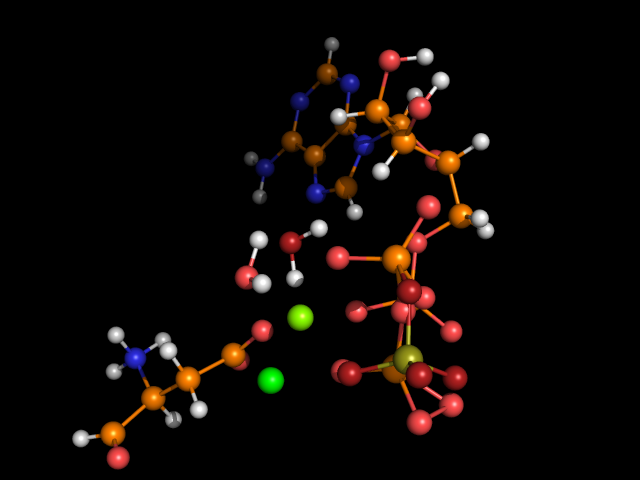
Как видно, положения воды, ионов магния и атомов γ-фосфата сильно отличаются в исходной и оптимизированной структурах. Атомы кислорода исходной структуры окрашены темно-красным, ион магния - ярко-зеленым, атом фосфора - оливковым. Нужно обратить внимание на то, что в оптимизированной структуре два атома кислорода у γ-фосфата расположены очень близко друг к другу (так близко, что PyMol изобразил ковалентную связь между ними). Такое в реальной структуре очень маловероятно, поэтому это, вероятно, артефакт оптимизации. Интересно расположение иона магния в оптимизированной структуре. Если внимательно посмотреть, можно заметить, что он находится примерно на равном расстоянии от трех атомов кислорода АТФ (α-, β- и γ-фосфатов), атома кислорода воды и одного из атомов кислорода боковой цепи аспартата. Это вполне может соответствовать реальности.
Сравним результат с записью 3PP1. Выделим в данной записи АТФ, ион магния, молекулу воды и остаток аспартата (208-й остаток цепи). Полученное изображение представлено ниже:
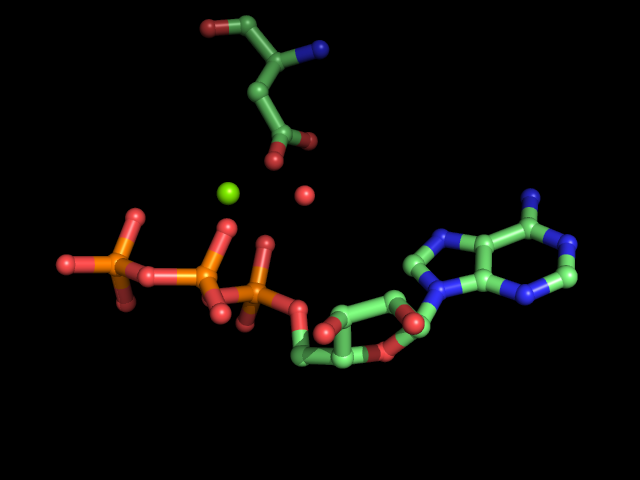
Наложим структуру записи на полученную оптимизированную структуру:
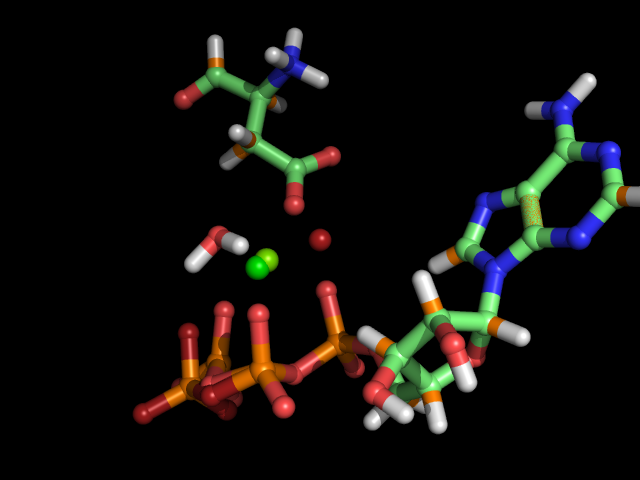
Видно, что в структуре записи 3PP1 ион магния координирует вокруг себя те же 4 атома, что и в оптимизированной структуре (и примерно на том же расстоянии). Однако, из наложения видно (в структуре записи 3PP1 атомы кислорода окрашены темно-красным, ион магния - ярко-зеленым), что молекула воды координируется в структуре записи 3PP1 с другой стороны (хотя, примерно, на том же расстоянии и под таким же углом), а остаток γ-фосфата немного повернут.
В целом, на мой взгляд, программа Mopac очень успешно справилась с поставленной задачей и предъявила достаточно правдоподобую структуру. Главный минус ее результата - наличие ковалентной связи между двумя атомами γ-фосфата (которые еще и отрицательно заряжены, поэтому никак не могут образовывать ковалентную связь).