С помощью babel сделаем pdb-файл этана et.pdb из результатов оптимизации из предыдущего практикума. Добавим путь к скриптам в системный путь:
export PATH=${PATH}:/home/preps/golovin/progs/bin
Теперь с помощью скрипта Ante_RED.pl подготовим pdb файл:
Ante_RED.pl et.pdbМультиплетность молекулы равна 1, заряд равен 0. Переименуем p2n-файл в Mol_red1.p2n и запустим RED:
RED-vIII.4.plНа выходе получаем файл Mol_m1-o1.mol2 с координатами атомов и зарядами.
В первых двух строчках зададим некоторые правила:
[ defaults ] ; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ 1 2 yes 0.5 0.8333Далее зададим типы атомов и собственно параметры для функции Леннорда-Джонса. Будем считать, что в случае этана Ван-дер-Ваальсовое взаимодействие между атомами углерода разных молекул минимально, так как углероды почти полностью экранированы атомами водорода. Поэтому поставим для углерода некоторые параметры. Ван-дер-Ваальсовый радиус водорода (sigma) известен. Получается, что в этом разделе имеем лишь одну переменную - epsilon для водорода:
[ atomtypes ] ; name at.num mass charge ptype sigma epsilon H 1 1.008 0.0000 A 1.06908e-01 1.00000e-00 C 6 12.01 0.0000 A 3.39967e-01 3.59824e-01Далее переходим непосредственно к описанию молекулы. Здесь мы описываем имя и указываем, что соседи через три связи не учитываются при расчете Ван-дер-Ваальсовых взаимодействий. Это верно, так как мы включаем это взаимодействие в торсионные углы:
[ moleculetype ] ; Name nrexcl et 3Добавим атомы этана:
[ atoms ]
; nr type resnr residue atom cgnr charge mass
1 C 1 ETH C1 1 -0.0189 12.01
2 C 1 ETH C2 2 -0.0155 12.01
3 H 1 ETH H1 3 0.0059 1.008
4 H 1 ETH H2 4 0.0059 1.008
5 H 1 ETH H3 5 0.0059 1.008
6 H 1 ETH H4 6 0.0056 1.008
7 H 1 ETH H5 7 0.0056 1.008
8 H 1 ETH H6 8 0.0056 1.008
Переходим к описанию связей. Константу жесткости и длину связи возьмем из предыдущего занятия:
[ bonds ]
; ai aj funct b0 kb
1 2 1 0.1554 150000.0
1 3 1 0.1085 180000.0
1 4 1 0.1085 180000.0
1 5 1 0.1085 180000.0
2 6 1 0.1085 180000.0
2 7 1 0.1085 180000.0
2 8 1 0.1085 180000.0
Переходим к описанию углов:
[ angles ]
; ai aj ak funct phi0 kphi
;around c1
3 1 4 1 109.500 200.400
3 1 5 1 109.500 200.400
4 1 5 1 109.500 200.400
3 1 2 1 109.500 200.400
4 1 2 1 109.500 200.400
5 1 2 1 109.500 200.400
;around c2
1 2 6 1 109.500 400.400
1 2 7 1 109.500 400.400
1 2 8 1 109.500 400.400
6 2 7 1 109.500 200.400
6 2 8 1 109.500 200.400
7 2 8 1 109.500 200.400
Переходим к торсионным углам:
[ dihedrals ]
; ai aj ak al funct t0 kt mult
3 1 2 6 1 0.0 0.62760 3
3 1 2 7 1 0.0 0.62760 3
3 1 2 8 1 0.0 0.62760 3
4 1 2 6 1 0.0 0.62760 3
4 1 2 7 1 0.0 0.62760 3
4 1 2 8 1 0.0 0.62760 3
5 1 2 6 1 0.0 0.62760 3
5 1 2 7 1 0.0 0.62760 3
5 1 2 8 1 0.0 0.62760 3
Теперь создадим список пар атомов, которые не должны считаться при расчете VdW. Особенность расчета 1-4 взаимодействий подразумевает, что в профиле торсионного угла участвует не только потенциал с cos, но и LJ отталкивание. Это удобно для точной параметризации, но нам пока не надо. Итак, добавляем список:
[ pairs ] ; ai aj funct 3 6 3 7 3 8 4 6 4 7 4 8 5 6 5 7 5 8Итак, основное описание молекулы создано. Теперь переходим к описанию системы:
[ System ] ; any text here first one [ molecules ] ;Name count et 38В результате получаем файл et.top с полным описанием системы.
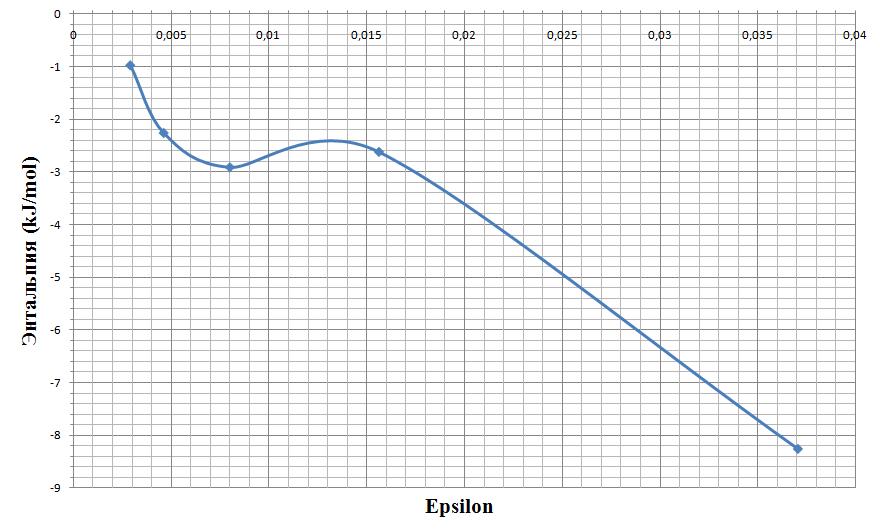
Тогда значение epsilon лежит в диапозоне примерно от 0.026 до 0.028.