Совмещение доменов цепей А записей 1HY0 и 1I0A в PyMOL
Получим из банка PDB записи 1HY0 и 1I0A и сохраним их в файлах 1HY0.pdb и 1I0A.pdb.На сервисе pDomains определим доменную структуру цепей А записей. Согласно CATH цепь А записи 1HY0 состоит из 3 доменов (координаты первого: 27-123, 195-234; второго: 124-194, 235-364, 437-463; третьего: 365-436). Цепь А записи 1I0A состоит из 3 доменов (координаты первого: 27-123, 195-234; второго: 124-194, 235-364, 437-463; третьего: 365-436).
Теперь мы можем построить в PyMOL командой align совмещения доменов цепей А из записей 1HY0 и 1I0A. Для этого по очереди выделим в PyMOL пары доменов из записей и воспользуемся командой:
align dom1, dom2Совмещение первых доменов представлено на картинке:

RMSD этого совмещения равно 0.371 (773 to 773 atoms), если совмещать все атомы, и 0.338 (117 to 117 atoms), если совмещать только Cα-атомы.
Совмещение вторых доменов представлено на картинке:

RMSD этого совмещения равно 0.409 (1371 to 1371 atoms), если совмещать все атомы, и 0.367 (200 to 200 atoms), если совмещать только Cα-атомы.
Совмещение третьих доменов представлено на картинке:

RMSD этого совмещения равно 0.527 (449 to 449 atoms), если совмещать все атомы, и 0.440 (67 to 67 atoms), если совмещать только Cα-атомы.
На рисунках зеленым окрашен остов цепи записи 1HY0, голубым - остов цепи записи 1I0A. Как видно из рисунков, пространственные структуры последовательностей очень похожи и прекрасно совпадают при наложении. Впрочем, между ними есть некоторые, пусть и небольшие, отличия. Например, на втором рисунке хорошо заметно несовпадение петель двух цепей (наверху картинки). Показатели rmsd также указывают на прекрасное совмещение доменов.
Построение структурного выравнивания цепей А записей 1HY0 и 1W0T
Построим программой PDBeFOLD структурное выравнивание цепи А из записи 1AKH и цепи А из записи 1W0T. Скачаем полученное выравнивание нажатием кнопки "Download aligned sequences (FASTA format)", перед этим отметив checkbox в таблице. Выравнивание было сохранено в файле 1akhalign.fasta.Затем по полученному выравниванию с помощью сервиса Geometrical core найдем геометрическое ядро с порогом 2 Å. Но прежде изменим названия последовательностей в fasta-файле в соответствии с требованиями этого сервиса. Отредактированное выравнивание было сохранено в файле 1akh1w0t.fasta. На выходе получаем список остатков (и позиций выравнивания), образующих геометрическое ядро. Этот список представлен в следующей таблице:
| Pos. | 1AKH_A | 1W0T_A |
| 11 | ALA83 | LYS389 |
| 12 | PHE84 | ASN390 |
| 13 | LEU85 | LEU391 |
| 15 | GLU87 | SER393 |
| 28 | LYS100 | TRP403 |
| 29 | GLU101 | SER404 |
| 30 | GLU102 | LYS405 |
| 31 | VAL103 | ILE406 |
| 41 | THR110 | THR416 |
| 42 | PRO111 | SER417 |
| 43 | LEU112 | VAL418 |
| 44 | GLN113 | MET419 |
| 45 | VAL114 | LEU420 |
| 46 | ARG115 | LYS421 |
| 47 | VAL116 | ASP422 |
| 48 | TRP117 | ARG423 |
| 49 | PHE118 | TRP424 |
| 50 | ILE119 | ARG425 |
| 51 | ASN120 | THR426 |
| 52 | LYS121 | MET427 |
| 53 | ARG122 | LYS428 |
| 54 | MET123 | LYS429 |
| 55 | ARG124 | LEU430 |
Наконец, совместим в PyMOL командой pair-fit структуры по Cα-атомам, входящим в геометрическое ядро. Важно, что на вход ей подаются два строго одинаковых множества атомов. Таким образом, получаем следующее изображение совмещенных структур:
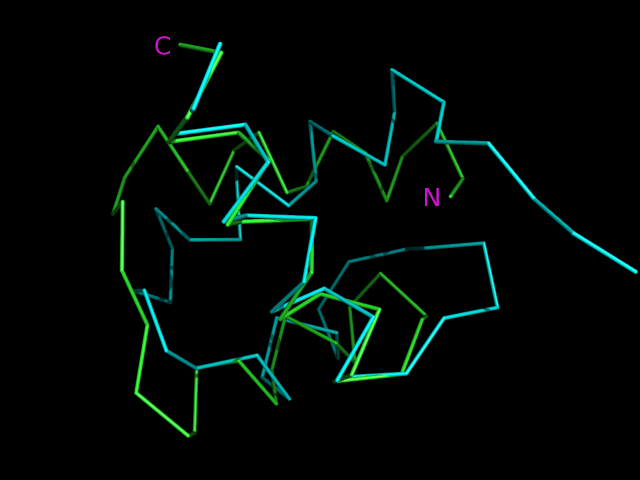
Зеленым на рисунке изображен остов цепи А записи 1AKH, а синим - остов цепи А записи 1W0T.
Как видно из рисунка, лучше всего выравнились (и, соответственно, совместились) альфа-спирали структур, расположенные на рисунке на переднем плане (примыкающие к С-концам цепей). В других участках цепей успешно выравнились отдельные остатки, не превышающие, однако, по длине 4 остатков (см. таблицу).
RMSD такого совмещения (по Сα-атомам, входящим в геометрическое ядро) = 0.865 (23 to 23 atoms). Это, конечно, превышает значение rmsd для совмещения доменов очень близких белков в предыдущем упражнении. Однако значение rmsd все равно очень хорошее, что говорит об успешном выравнивании и совмещении структур.
Для сравнения посмотрим на результат команды align на полных цепях. Соответствующий рисунок представлен ниже:

Очевидно, что это выравнивание категорически не верно. RMSD такого совмещения = 6.198 (161 to 161 atoms). Это уже очень большая величина, не идущая в сравнение ни с одним предыдущим rmsd.
Теперь сравним два полученных результата с результатом экспериментальной команды super. Для этого подадим ей на вход два множества (две цепи), а затем применим следующую команду:
super ch1, ch2В результате было получено следующее изображение:
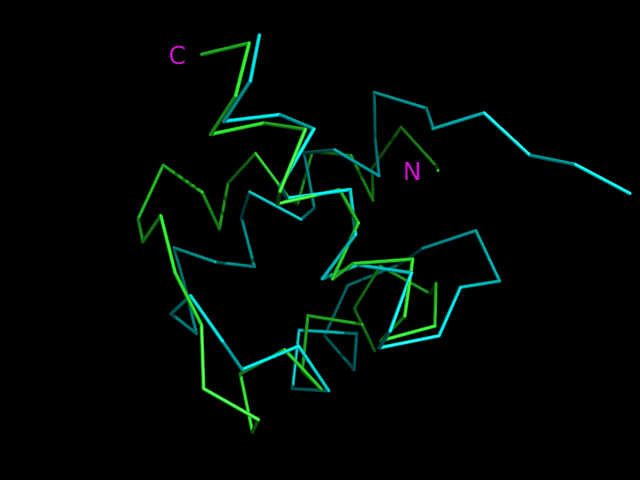
RMSD полученного выравнивания равно 2.186 (192 to 192 atoms). Это значение rmsd куда хуже значения, полученного с помощью программ SSM и pair-fit. Впрочем, из рисунка видно, что структуры совместились почти также, как и в результате выравнивания программой SSM. Таким образом, результат программы super достаточно достоверен.
Если подать на вход программе только Cα-атомы структур, цепи совместятся точно так же, но rmsd такого совмещения будет равно 1.97 (39 to 39 atoms). Скорее всего, высокие значения rmsd для этих выравниваний объясняются прежде всего тем, что в этом случае совмещались все остатки цепей, а в случае программы pair-fit - только остатки, входящие в геометрическое ядро.