Поиск гипотетических гомологов белка DPS_ECOLI в разных БД
| Поиск по БД Swiss-Prot | Поиск по БД PDB | Поиск по БД "nr" | |
| 1. Лучшая находка (в принципе должна соответствовать заданному белку) | |||
| Идентификатор БД | DPS_ECOLI | 1F30 | YP_688332.1 |
| E-value | 9.10-93 | 10-94 | 10-92 |
| Вес (в битах) | 340 | 340 | 341 |
| % идентичности | 100% | 100% | 100% |
| Найдены ли другие белки с теми же значениями E-value и веса в битах? Если найдены, то укажите общее число и приведите один идентификатор (любой, но желательно Swiss-Prot ID) |
Найдено еще 11 белков с теми же значениями E-value и тем же весом в битах. Один из них - DPS_KLUCI | Кроме найденных структур 12 полипептидных цепей моего белка найдены структуры 12 полипептидных цепей белка 1F33 с таким же E-value и весом выравнивания (на самом деле, это тот же белок DPS_ECOLI, просто связанный с другими лигандами) | Найден еще один белок с таким же значением E-value и тем же весом в битах - ABF03027.1 |
| 2. Сколько хороших кандидатов в гомологи найдено? (число находок в списке описаний, Descriptions, с E-value < 1E-10) | 24 находки с E-value, не превышающим 10-10. Лучшая находка - DPS_KLUCI, "худшая из лучших" - DPS_STRMU | 9 находок с E-value, не превышающим 10-10. Лучшая находка - 1F30, "худшая из лучших" - 2IY4 | 149 находок с E-value, не превышающим 10-10. Лучшая находка - YP_688332.1, "худшая из лучших" - NP_816831.1 |
| 2. "Худшая" находка (последняя в выдаче с E-value < 1) | |||
| Номер находки в списке описаний (Descriptions) | 46 | 31 | 769 |
| Идентификатор БД | DPS_HALSA | �2C41 | YP_001663126.1 |
| E-value | 0.047 | 0.98 | 0.98 |
| Вес (в битах) | 37.0 | 28.9 | 36.2 |
| % идентичности | 25% | 22% | 26% |
| % сходства | 45% | 43% | 47% |
| Длина выравнивания | 118 | 144 | 110 |
| Координаты выравнивания (номера первых и последних а.о.) | В DPS_ECOLI с 1-ого по 115-й; в DPS_HALSA с 1-го по 116-й | В 1F30 с 26-го по 163-й; в 2C41 с 8-го по 147-ой | В DPS_ECOLI c 54-го по 157-ой; в YP_001663126.1 с 61-го по 160-ый |
| % гэпов | 4% | 6% | 14% |
Итак, изучаемый белок (DPS_ECOLI) удалось найти и в SwissProt, и в "nr", однако в "nr" меня удивило то, что кроме него был найдено еще два белка с процентом идентичности, равным также 100%, но для которых вес (в битах) был на 1 больше, чем для самого изучаемого белка (!!!) и E-value меньше, чем для изучаемого белка (!!!). В PDB была найдена структура всех 12 полипептидных цепей моего белка.
Параметры выравнивания белка с самим собой при поиске по разным БД одинаковые, за исключением E-value. Это объясняется тем, что E-value ищется по формуле E = l.K.L.e-λS, где l - длина последовательности, S - вес последовательности, L - объем банка данных, а λ и K - параметры системы подсчета и размера поискового пространства. Таким образом, как видно из формулы, чем больше объем данных, тем больше E-value. Именно поэтому самый большой E-value в nr (для выравнивания белка с самим собой он составляет 10-92), так как эта БД - самая большая; меньше в курируемой БД SwissProt (9.10-93), ну и, наконец, меньше всего E-value в PDB (10-94), так как эта БД самая маленькая из всех, ведь получение третичной структуры белка - процесс очень трудоемкий и долгий, условия для кристаллизации подбираются месяцами и даже годами.
Число потенциальных гомологов при поиске в различных БД различается по той же самой причине (из-за размера различных баз данных). В "nr" найдено 149 находок, ведь эта БД объединяет в себе и PDB, и SwissProt, и много других БД; меньше находок найдено в SwsissProt (24 находки); меньше всего находок - в PDB (9 находок).
"Худшие" находки в различных БД не совпадают. Это объясняется, опять же, размером БД. PDB - самая маленькая из данных баз данных, поэтому, если какой-либо белок имеет в ней E-value, равный 0,98, то в остальных БД (SwissProt и, тем более, "nr") E-value этого же белка будет значительно больше (больше 1), поэтому в предел меньше 1 этот белок просто не попадет. Из "худших" находок самая лучшая - DPS_HALSA из SwissProt (его вес в битах равен 37.0). Казалось бы, лучшим должен был бы быть "худший" белок из БД "nr", ведь его E-value в остальных БД будет еще меньше, а значит больше будет и вес. В данном случае это не так, потому что, как уже было сказано, БД "nr" очень большая, и многие белки, содержащиеся в этой БД, в SwissProt и, тем более, Pdb не содержатся. Поэтому лучшая находка из "худших" найдена в SwissProt, в "nr" ее E-value равен около 0,5.
Поиск гипотетических гомологов изучаемого белка (DPS_ECOLI) с фильтром по таксонам в БД SwissProt
| Поиск в таксоне Homo sapiens (taxid:9606) | Поиск в таксоне Archaea (taxid:2157) | Поиск в таксоне Actinobacteria (class) (taxid:1760) | Поиск в таксоне Alteromonadales (taxid:135622) | Поиск в таксоне Vibrionaceae (taxid:641) | |
| Идентификатор БД | Нет | Нет | DPS_MYCS2 | Нет | Нет |
| E-value (<=0.001) | - | - | 7.10-19 | - | - |
| Вес (в битах) | - | - | 88.2 | - | - |
| % идентичности | - | - | 35% | - | - |
| % сходства | - | - | 51% | - | - |
| Длина выравнивания | - | - | 154 | - | - |
| Координаты выравнивания (номера первых и последних а.о.) | - | - | В DPS_ECOLI с 16-го по 167-ой; в DPS_MYCS2 с 4-го по 157-ой | - | - |
| % гэпов | - | - | 1% | - | - |
Лучший гомолог был найден в отделе Актинобактерий. Это белок - DPS_MYCS2 (кстати, именно этот белок был дан мне для выполнения заданий по локальному и глобальному выравниванию). Принадлежит он бактерии Mycobacterium smegmatis str. MC2 155 (систематика: Bacteria; Actinobacteria; Actinobacteridae; Actinomycetales; Corynebacterineae; Mycobacteriaceae; Mycobacterium).
Просмотрев информацию по данному белку, понимаю, что он действительно, скорее всего, является гомологом (ортологом): он выполняет такую же функцию, имеет очень похожее строение на DPS_ECOLI. В семействе Vibrionaceae, порядке Alteromonadales, царстве Архей и виде Homo sapiens возможных гомологов найдено не было. Для таксонов низкого ранга (семейство Vibrionaceae и порядок Alteromonadales) это объясняется, скорее всего, тем, что у них просто нет гомологов этого белка, соответствующую функцию выполняет белок совершенно другого происхождения (кроме того, возможно, что он выполняет не только эту функцию). Кроме того, возможно, что эти таксоны были изучены не достаточно полно, и гомолог белка DPS_ECOLI у них есть, но пока не найден.
Для таксонов более высокого ранга (царство Archea и вид Homo sapiens) отсутствие находок можно объяснить, во-первых, тем, что у них просто нет гомологов данного белка (либо не возникает проблемы, которую решает данный белок, либо ее решает белок, имеющий другое происхождение). Кроме того, в случае человека, возможно, что у нас в клетках (скорее всего в митохондриях) содержится белок, имеющий какое-то отдаленное происхождение с DPS_ECOLI, но естественно за такой грандиозный эволюционный путь он так сильно видоизменился, что стал просто неузнаваем по сравнению с DPS_ECOLI. Поэтому вес выравнивания его с DPS_ECOLI, естественно, стал совсем небольшим, и этого белка BLASTP не выдал в качестве претендента на гомолога DPS_ECOLI.
Поиск белка по его фрагменту
На первом занятии мне был дан следующий фрагмент белка в fasta-формате:
>seq2 SSSKLLKVYTRNNLDPAVС помощью BLASTP я нашел белок, из которого был дан фрагмент. Это белок DPS_ACIAD (его AС - Q6FCX7), его последовательность в fasta-формате:
>gi|81613258|sp|Q6FCX7.1|DPS_ACIAD RecName: Full=DNA protection during starvation protein MSKSSSKLLKVYTRNNLDPAVKESTVKILNQILANLIDLSLLTKQAHWNMRGSNFIAVHEMLDTFRTSLI THLDNVAERAVQIGGTALGTTQTVSTTSQLSAYPVEIHNVQDHLKELADRYGIVANHLRDTIEEIQDPIS EDIIHAALEDLDQYLWFLEANIEQDKTTA
Результаты поиска белка DPS_ECOLI в Swiss-Prot по фрагменту последовательности
| Поиск по фрагменту | Поиск по полной последовательности |
|
| АС лучшей находки | Q6FCX7 | Q6FCX7 |
| E-value | 4.10-9 | 7.10-95 |
| Вес (в битах) | 58.3 | 345 |
| Найдены ли другие белки с теми же значениями E-value и веса в битах? |
Нет, E-value следующего по поиску белка составляет уже 2.4 (вес - 29.1) | Нет |
Вес выравнивания при поиске по полной последовательности во много раз больше веса при поиске по фрагменту. Это объясняется длиной выравниваний (естественно, длина полной последовательности будет значительно больше, а значит больше и вес выравнивания).
E-value выравнивания при поиске по полной последовательности в 86 (!!!) раз меньше E-value при поиске по фрагменту. Это можно легко объяснить, вспомнив формулу вычисления E-value. Напомню, что E = l.K.L.e-λS, где l - длина последовательности, S - вес последовательности, L - объем банка данных, а λ и K - параметры системы подсчета и размера поискового пространства. Сейчас нам важны именно l и S. Как видно, чем больше длина последовательности, тем больше E-value. Поэтому, казалось бы, E-value выравнивания по полной последовательности должно быть больше E-value выравнивания по фрагменту. Но ведь чем больше вес выравнивания, тем меньше E-value, причем вес выравнивания берется в степени, а это значительно сильнее влияет на функцию E, чем линейная зависимость длины последовательности. Таким образом, E-value выравнивания при поиске по полной последовательности получается значительно меньше E-value выравнивания при поиске по фрагменту.
Теперь сравним выравнивание, выданное BLASTP, с выравниванием, сделанным мной с помощью GeneDoc. Так выглядит мое пробное выравнивание из первого занятия:
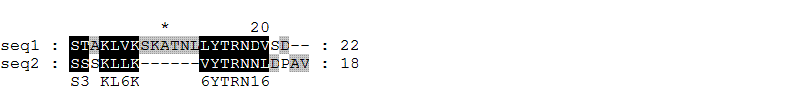
А так выглядит выравнивание полных последовательностей белка DPS_ECOLI и DPS_ACIAD, сделанное с помощью BLASTP (соответствующие участки выделены более крупным шрифтом):
Вес = 201 бит (510)
E-value = 7.10-57
Идентичность = 100/166 (60%)
Сходство = 133/166 (80%)
Гэпы = 6/166 (3%)
DPS_ECOLI 2 STAKLVKSKATNLLYTRNDVSDSEKKATVELLNRQVIQFIDLSLITKQAHWNMRGANFIA 61
S++KL+K +YTRN++ + K++TV++LN+ + IDLSL+TKQAHWNMRG+NFIA
DPS_ACIAD 4 SSSKLLK------VYTRNNLDPAVKESTVKILNQILANLIDLSLLTKQAHWNMRGSNFIA 57
DPS_ECOLI 62 VHEMLDGFRTALIDHLDTMAERAVQLGGVALGTTQVINSKTPLKSYPLDIHNVQDHLKEL 121
VHEMLD FRT+LI HLD +AERAVQ+GG ALGTTQ +++ + L +YP++IHNVQDHLKEL
DPS_ACIAD 58 VHEMLDTFRTSLITHLDNVAERAVQIGGTALGTTQTVSTTSQLSAYPVEIHNVQDHLKEL 117
DPS_ECOLI 122 ADRYAIVANDVRKAIGEAKDDDTADILTAASRDLDKFLWFIESNIE 167
ADRY IVAN +R I E +D + DI+ AA DLD++LWF+E+NIE
DPS_ACIAD 118 ADRYGIVANHLRDTIEEIQDPISEDIIHAALEDLDQYLWFLEANIE 163
Как видно, выравнивания совпадают.
Сравнение выравниваний, выданных программой BLASTP, с оптимальными глобальным и локальным выравниваниями.
Получим оптимальное глобальное и локальное выравнивания белков DPS_ACIAD и DPS_ECOLI с помощью программ needle и water соответственно. Но в BLASTP за открытие пропуска штраф - 11.0, а за продолжение - 1.0, поэтому поставим для программ needle и water следующие параметры: штраф за открытие пропуска - 11.0, штраф за продолжение пропуска - 1.0.
Итак, выравнивание BLASTP:
Вес = 201 бит (510)
E-value = 7.10-57
Идентичность = 100/166 (60%)
Сходство = 133/166 (80%)
Гэпы = 6/166 (3%)
DPS_ECOLI 2 STAKLVKSKATNLLYTRNDVSDSEKKATVELLNRQVIQFIDLSLITKQAHWNMRGANFIA 61
S++KL+K +YTRN++ + K++TV++LN+ + IDLSL+TKQAHWNMRG+NFIA
DPS_ACIAD 4 SSSKLLK------VYTRNNLDPAVKESTVKILNQILANLIDLSLLTKQAHWNMRGSNFIA 57
DPS_ECOLI 62 VHEMLDGFRTALIDHLDTMAERAVQLGGVALGTTQVINSKTPLKSYPLDIHNVQDHLKEL 121
VHEMLD FRT+LI HLD +AERAVQ+GG ALGTTQ +++ + L +YP++IHNVQDHLKEL
DPS_ACIAD 58 VHEMLDTFRTSLITHLDNVAERAVQIGGTALGTTQTVSTTSQLSAYPVEIHNVQDHLKEL 117
DPS_ECOLI 122 ADRYAIVANDVRKAIGEAKDDDTADILTAASRDLDKFLWFIESNIE 167
ADRY IVAN +R I E +D + DI+ AA DLD++LWF+E+NIE
DPS_ACIAD 118 ADRYGIVANHLRDTIEEIQDPISEDIIHAALEDLDQYLWFLEANIE 163
Оптимальное глобальное выравнивание в программе needle:
Длина: 175
Идентичность: 100/175 (57.1%)
Сходство: 133/175 (76.0%)
Гэпы: 14/175 ( 8.0%)
Вес: 521.0
DPS_ECOLI 1 --MSTAKLVKSKATNLLYTRNDVSDSEKKATVELLNRQVIQFIDLSLITK 48
.|::||:| :||||::..:.|::||::||:.:...|||||:||
DPS_ACIAD 1 MSKSSSKLLK------VYTRNNLDPAVKESTVKILNQILANLIDLSLLTK 44
DPS_ECOLI 49 QAHWNMRGANFIAVHEMLDGFRTALIDHLDTMAERAVQLGGVALGTTQVI 98
||||||||:||||||||||.|||:||.|||.:||||||:||.||||||.:
DPS_ACIAD 45 QAHWNMRGSNFIAVHEMLDTFRTSLITHLDNVAERAVQIGGTALGTTQTV 94
DPS_ECOLI 99 NSKTPLKSYPLDIHNVQDHLKELADRYAIVANDVRKAIGEAKDDDTADIL 148
::.:.|.:||::|||||||||||||||.||||.:|..|.|.:|..:.||:
DPS_ACIAD 95 STTSQLSAYPVEIHNVQDHLKELADRYGIVANHLRDTIEEIQDPISEDII 144
DPS_ECOLI 149 TAASRDLDKFLWFIESNIE------ 167
.||..|||::|||:|:|||
DPS_ACIAD 145 HAALEDLDQYLWFLEANIEQDKTTA 169
Как видно, отличаются эти два выравнивания лишь длиной выравнивания. В выравнивании BLASTP не хватает 3 колонок в начале и 6 колонок в конце (которые имеются в оптимальном глобальном выравнивании программы needle) (эти колонки выделены крупным шрифтом в выравнивании needle).
В связи с этим у двух выравниваний различные проценты идентичности и сходства. Процент идентичности в выравнивании BLASTP равен 60%, а в оптимальном глобальном выравнивании программы needle - 57,1%. Процент сходства в выравнивании BLASTP равен 80%, а в оптимальном глобальном выравнивании needle - 76%.
В диапозоне от 2-ого до 167-ого аминокислотного остатка у DPS_ECOLI и от 4-го до 163-го аминокислотного остатка у DPS_ACIAD сопоставления в двух выравниваниях одни и те же.
Вес выравнивания BLASTP составляет 510, вес оптимального глобального выравнивания needle - 520 (именно потому что длина выравнивания в needle больше длины выравнивания BLASTP).
Теперь рассмотрим оптимальное локальное выравнивание с помощью программы water:
Длина: 166
Идентичность: 100/166 (60.2%)
Сходство: 133/166 (80.1%)
Гэпы: 6/166 ( 3.6%)
Вес: 522.0
DPS_ECOLI 2 STAKLVKSKATNLLYTRNDVSDSEKKATVELLNRQVIQFIDLSLITKQAH 51
|::||:| :||||::..:.|::||::||:.:...|||||:|||||
DPS_ACIAD 4 SSSKLLK------VYTRNNLDPAVKESTVKILNQILANLIDLSLLTKQAH 47
DPS_ECOLI 52 WNMRGANFIAVHEMLDGFRTALIDHLDTMAERAVQLGGVALGTTQVINSK 101
|||||:||||||||||.|||:||.|||.:||||||:||.||||||.:::.
DPS_ACIAD 48 WNMRGSNFIAVHEMLDTFRTSLITHLDNVAERAVQIGGTALGTTQTVSTT 97
DPS_ECOLI 102 TPLKSYPLDIHNVQDHLKELADRYAIVANDVRKAIGEAKDDDTADILTAA 151
:.|.:||::|||||||||||||||.||||.:|..|.|.:|..:.||:.||
DPS_ACIAD 98 SQLSAYPVEIHNVQDHLKELADRYGIVANHLRDTIEEIQDPISEDIIHAA 147
DPS_ECOLI 152 SRDLDKFLWFIESNIE 167
..|||::|||:|:|||
DPS_ACIAD 148 LEDLDQYLWFLEANIE 163
Это выравнивание ничем не отличается от выравнивания BLASTP. Выравнивания абсолютно идентичны, однако проценты идентичности, сходства и гэпов посчитаны с разной точностью, поэтому процент идентичности, сходства и гэпов оптимального локального выравнивания water отличается от процентов BLASTP: процент идентичности равен 60.2%, процент сходства - 80.1%, процент гэпов - 3.6% (в выравнивании BLASTP проценты округлялись до целых).
Особое внимание я обратил на весы этих выравниваний. Выравнивания абсолютно идентичны, значит и вес должен быть у них один и тот же, однако почему-то разница в весах оптимального локального выравнивания и выравнивания BLASTP вполне внушительная (составляет 12). Это довольно странно, ведь параметры мы ставили одинаковые. Причем, если сравнить весы оптимального локального и оптимального глобального выравнивания water и needle, все кажется правильным (ведь отличаются они на 1, а вес пары M-K как раз составляет -1). Вначале мне пришла в голову следующая версия объяснения такого "феномена": в BLASTP, во-первых, штраф за продолжение пропуска на каждый пробел распространяется и на первый пробел пропуска (то есть за первый пробел пропуска штраф равен 12, а не 11), а во-вторых, штраф за открытие пропуска ставится так же с противоположной стороны пропуска (то есть штраф за открытие пропуска начисляется двум крайним пробелам). Тогда разница как раз и составит 1 + 11 = 12. Но, как оказалось на самом деле, эта версия оказалась ошибочной (хотя в данном случае она оказалась вполне применимой). На самом деле BLASTP действительно начисляет штраф за продолжение пропуска на первый пробел пропуска (то есть за первый пробел штраф равен 12, а не 11), но с противоположной стороны штраф за открытие пропуска он не начисляет. Просто BLASTP при начислении штрафов и бонусов за сопоставляемые буквы использует немного другую матрицу, чем привычную нам BLOSUM62. BLASTP использует немного модифицированную матрицу с соответственно другими значениями штрафов и бонусов за сопоставляемые буквы. Поэтому суммарный вес выравнивания немного отличается от веса идентичного выравнивания, но полученного с помощью программ water и needle.
Такая минимальная разница между тремя выравниваниями (заключающаяся только в том, что оптимальное локальное выравнивание water и выравнивание BLASTP являются фрагментом оптимального глобального выравнивания needle) говорит о том, что это выравнивание и вправду является самым оптимальным, и скорее всего эволюция белков пошла именно в таком направлении (при образовании гомологов).