 |
 |
Учебный сайт Морозова Александра |
<< Назад к странице 3 семестра
Практикум 11Часть I: подготовка чтенийЗадания №1-2. Анализ качества чтений до и после очистки.
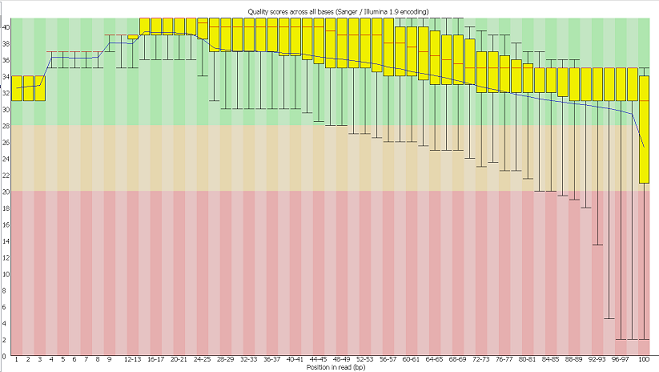 Рис. 1. График FastQC "Per base quality" до чистки 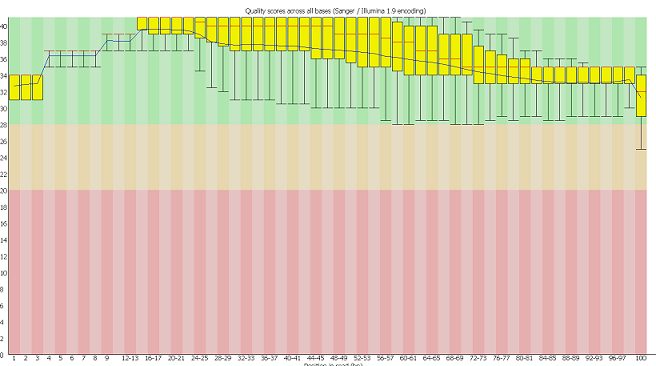 Рис. 2.График FastQC "Per base quality" после чистки Число чтений до очистки:11427 Число чтений после очистки:11091 После выполнения первой команды были удалены все нуклеотиды с качеством ниже 20. При этом часть ридов стала короче. После выполнения второй команды были удалены все риды длиной меньше 50. Часть II: картирование чтений1) Индексирование референсной последовательности:/home/students/y06/anastaisha_w/hisat2-2.0.5/hisat2-build chr22.fasta chrbuild 2) Получение выравнивания прочтений и референса в формате .sam/home/students/y06/anastaisha_w/hisat2-2.0.5/hisat2 -x chrbuild -U trimmed_chr22_1.fastq --no-spliced-alignment --no-softclip > align.sam 3) Перевод файла align в формат .bam:samtools view align.sam -bo align.bam 4) Отсортировать выравнивание чтений с референсом по координате в референсе начала чтенияsamtools sort align.bam -T temp.txt -o sort_align.bam 5) Индексирование отсортированного .bam файлаsamtools index sort_align.bam 6) Получение файла со статистикой:samtools stats sort_align.bam > stats.txt Из этого файла видно, что 11061 чтений было откартировано на хромосому, 54 - не картировано. Часть III: Поиск SNP и инделей.1) Создание файла с полиморфизмами в формате .bcf:samtools mpileup -uf chr22.fasta sort_align.bam > snp.bcf 2) Создание файла со списком отличий между референсом и чтениями в формате .vcfbcftools call -cv snp.bcf > snp.vcf
Всего получено 225 SNP, из них инделей - 10 Аннотация SNP1) Создание файла, совместимого с программой annovar:perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 /nfs/srv/databases/ngs/alex_morozov/pr11/snp.vcf > /nfs/srv/databases/ngs/alex_morozov/pr11/snp.annovar Refgene1) Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -out /nfs/srv/databases/ngs/alex_morozov/pr11/rs.refgene -build hg19 /nfs/srv/databases/ngs/alex_morozov/pr11/snp.annovar /nfs/srv/databases/annovar/humandb/ 2) Содержание SNP в различных участках генома: Экзоны - 25 Интроны - 197 Таким образом, очевидно, что наибольшее число SNP накапливается в интронах Dbsnp1) Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out /nfs/srv/databases/ngs/alex_morozov/pr11/dbsnp.snp -build hg19 -dbtype snp138 /nfs/srv/databases/ngs/alex_morozov/pr11/snp.annovar /nfs/srv/databases/annovar/humandb/ 2) 177 snp имеют rs 48 snp rs не имеют Clinvar1) Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out /nfs/srv/databases/ngs/alex_morozov/pr11/rs.clinvar -dbtype clinvar_20150629 -buildver hg19 /nfs/srv/databases/ngs/alex_morozov/pr11/snp.annovar /nfs/srv/databases/annovar/humandb/ 2) Получил на выход 2 файла. В одном были перечислены snp, имеющие влияние на здоровье носителя. Всего таких snp в файле было 2. В другом файле были перечислены snp без аннотации 1000Genomes1) Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out /nfs/srv/databases/ngs/alex_morozov/pr11/rs.1000genomes -buildver hg19 -dbtype 1000g2014oct_all /nfs/srv/databases/ngs/alex_morozov/pr11/snp.annovar /nfs/srv/databases/annovar/humandb/ 2) По аннотации данной БД можо приблизительно оценить частоту встречаемости SNP.
Самая высокая частота встречаемости SNP из числа исследуемых в этом практикуме: 0.9998, самая низкая частота встречаемости: 0.00319489 GWAS1) Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out /nfs/srv/databases/ngs/alex_morozov/pr11/rs.gwas -build hg19 -dbtype gwasCatalog /nfs/srv/databases/ngs/alex_morozov/pr11/snp.annovar /nfs/srv/databases/annovar/humandb/ 2) По аннотации GWAS можно узнать, к каким нуклеотидным и аминокислотным заменам привели snp. Полученный файл, где должны находится аннотированные записи, оказался пустым, следовательно, snp не приводят к фенотипическим изменениям |
|||||||||||||||||||||||||||||||||||||||||||