 |
 |
Учебный сайт Морозова Александра |
<< Назад к странице 3 семестра
Практикум 12Часть I: подготовка чтений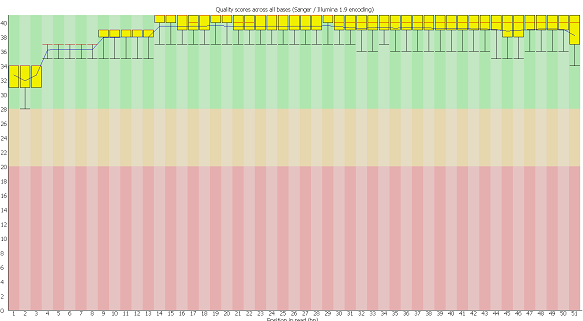 Рис. 1.Анализ качества чтений Как можно видеть на изображении, полученном с помощью программы FastQC для Windows, данные чтения имеют хорошее качество и приемлимую длину, поэтому не нуждаются в чистке. 1) Индексирование референсной последовательности: /home/students/y06/anastaisha_w/hisat2-2.0.5/hisat2-build chr22.fasta chrbuild 2) Выравнивание прочтений и референса: /home/students/y06/anastaisha_w/hisat2-2.0.5/hisat2 -x chrbuild -U chr22.1.fastq --no-softclip > align.sam * В отличие от практикума 11, в этом коде нет параметра "--no-spliced-alignment", т.к. работа ведется с последовательностями РНК, прошедших сплайсинг и, возможно, имеющих разрывы. 3) Перевод выравнивания в бинарный код (.bam): samtools view align.sam -bo align.bam 4) Сортировка выравнивания: samtools sort align.bam -T temp.txt -o sort_align.bam 5) Индексирование отсортированного файла samtools index sort_align.bam 6) Получение файла со статистикой: samtools stats sort_align.bam > stats.txt Согласно данным полученного файла, всего из 24294 прочтений картировались 23927; не картированными остались 367. Часть II: подcчет чтений1) Перевести файл с выравниванием из .bam формата в .bed формат: /P/y14/term3/block4/SNP/bedtools2/bin/bedtools bamtobed -i sort_align.bam > sort_align.bed 2)Подсчет чтений: bedtools intersect -a /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed -b sort_align.bed -u > gene1.bed Данная команда позволила мне узнать, какие гены покрывают данные чтения, через пересечения с общим файлом с разметкой генов. Опция -u позволила мне получить только те чтения, которые хотя бы раз пересекались с генами. При запуске без этого параметра полученный файл был гораздо больше и бессмысленней.
По результатам работы данной программы, большая часть чтений легла на ген белка PRAME (193). Небольшая часть легла на ген LL22NC03-63E9.3 (2), также кодирующий белок. |