Гомологичное моделирование комплекса белка с лигандом
В данном задании я работала с белком лизоцимом из указанного организма: LYS_ASTRU.
Данный белок является неполным и его последовательность в Uniprot представлена лишь 25-ю а.о., поэтому при выравнивании такого фрагмента белка с белком лизоцима форели 1LMP, получилось вот такое
выравнивание:

1) Выравнивание было сохранено в формате PIR.
2) Далее файл формата PIR был исправлен:
Переименовала последовательность в файле выравнивания:
| Было |
Стало |
| >P1;uniprot|P37712|LYSC_CAMDR |
>P1;seq |
| >P1;1LMP|PDBID|CHAIN|SEQUENCE |
>P1;1lmp |
После имени последовательности моделируемого белка надо добавить строчку:
sequence:ХХХХХ::::::: 0.00: 0.00
эта строчка описывает входные параметры последовательности для modeller. После имени последовательности белка-образца добавить:
structureX:1lmp_edited.ent:1 :A: 132 :A:undefined:undefined:-1.00:-1.00
эта строчка описывает, какой файл содержит структуру белка с этой последовательностью,
номера первой и последней аминокислот в структуре, идентификатор цепи и т.д.
В конце каждой последовательности надо добавить символы:
/.
Символ "/" означает конец цепи белка. Точка указывает на то, что имеется один лиганд (если бы было два лиганда стояли бы две точки).
Получилось модифицированное выравнивание в формате PIR.
3) Файл со структурой был также изменен.
Пример:
| Было |
Стало |
| HETATM 1014 O7 NAG 130 |
HETATM 1014 O7A NAG 130 |
| HETATM 1015 C1 NAG 131 |
HETATM 1015 C1B NAG 130 |
Измененный файл был сохранен как 1lmp_edited.ent.
4) Создание скрипта:
lys_astru.py
В скрипте указано:
что нужно использовать стандартные валентные углы в полипептидной цепи (строчка 4);
что дополнительно нужно сохранять взаимное расположение определенных пар атомов (3.5 ангстрема);
атомы белка, образующие водородные связи с тремя атомами лиганда - строчки 5-7 с ID пар атомов (длина данного белка 226 а.о., а белка-модели - 25,
поэтому номера а.о. лиганда сильно изменились);
параметры взаимного расположения атомов пары описаны в строчке 9-10. 3 точки могут однозначно расположить сложную структуру в пространстве,
поэтому мы выбираем водородные связи как источник данных точек;
что ковалентные связи в гетероатомах нужно вычислять по расстояниям между атомами, строчка 12;
что имя файла с выравниванием и имена последовательностей образца и моделируемого белка, строчка 13 (а имя файла со структурой содержится в выравнивании);
что число и номера моделей, которые нужно построить (в данном примере 5 моделей), строки 14-15
что пора строить модель, строчка 16
Запустим исполнение скрипта командой
Получили вот такие модели (5шт):
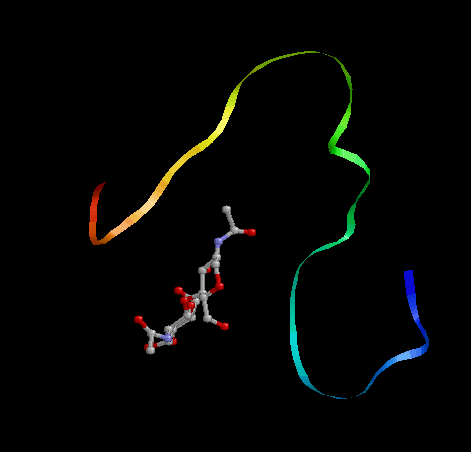
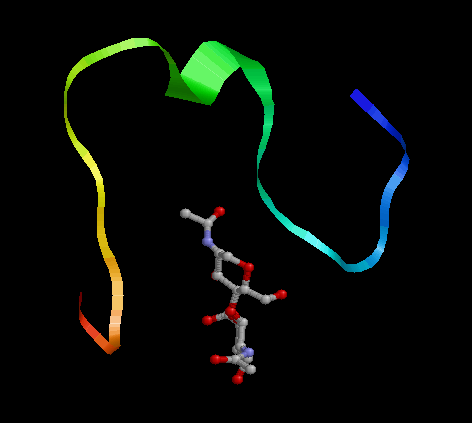

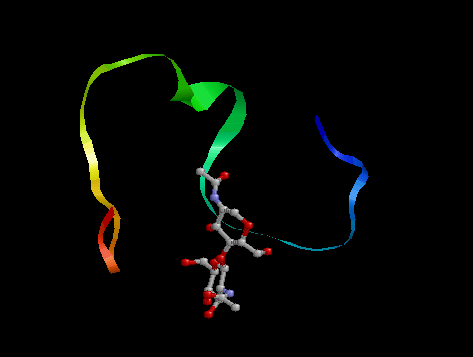
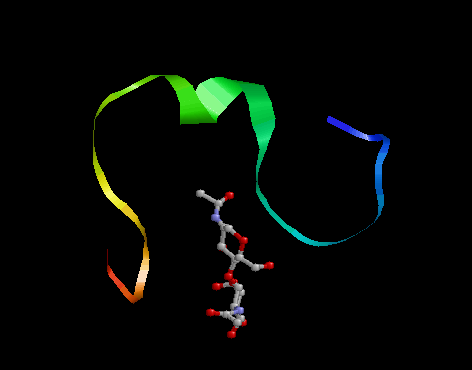
Сравнивать полученные модели с исходной, думаю, бессмысленно, хотя мой белок в приципе напоминает некий набросок полного белка.
5)Теперь необходимо проверить правильность такой модели:
Я выбрала следующие критерии для оценки лучшей модели:
1) Ramachandran Z-score
1 - (-4.991)
2 - (-4.878)
3 - (-4.347)
4 - (-2.195)
5 - (-2.374)
1LMP - (-0.965)
2) Anomalous bond lengths
1: RMS Z-score for bond lengths: 1.114
RMS-deviation in bond distances: 0.023
2: RMS Z-score for bond lengths: 1.120
RMS-deviation in bond distances: 0.023
3: RMS Z-score for bond lengths: 1.118
RMS-deviation in bond distances: 0.023
4:RMS Z-score for bond lengths: 1.101
RMS-deviation in bond distances: 0.023
5: RMS Z-score for bond lengths: 1.085
RMS-deviation in bond distances: 0.022
1LMP: RMS Z-score for bond lengths: 0.839
RMS-deviation in bond distances: 0.023
Наиболее близкими по этим параметрам к 1LMP оказались модели 4 и 5: 4ая ближе по Ramachandran Z-score,
зато по RMS Z-score for bond lengths ближе 5ая модель. Но по обоим параметрам они несильно отличаются между
собой, поэтому можно выбрать, к примеру 4ую модель для дальнейшего исследования.
© Шерстюк Александра, MSU 2009