Попробуем разные варианты BLAST для короткого контига из сборки генома организма, с которым работали на прошлом практикуме. С помощью программы infoseq из пакета EMBOSS посмотрим, скэффолды какой длины присутствуют в сборке. Так как все скэффолды данной сборки гораздо больше 10 000 п.н., будем работать со скэффолдом, имеющем идентификатор NW_023397510. Его длина составляет 156074 п.о
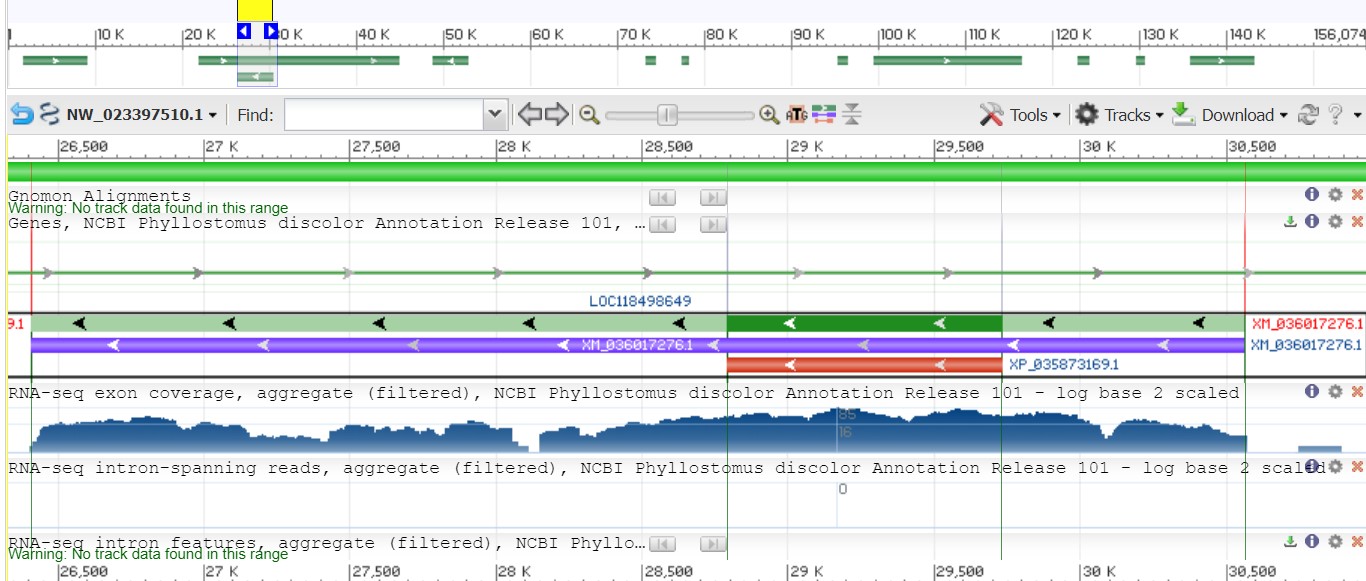
Рассмотрим схему расположения генов в этом скэффолде (Рис. 1). На верхней панели видно, что в нём 11 генов (они обозначены зелёным). Дальше будем работать с фрагментом этого скэффолда с координатами [26409:30563]. На схеме (Рис.1) видим, что зелёным отмечен сам ген, светло-зелёным — 5'- и 3'-нетранслируемые области, фиолетовым — соответствующая мРНК и красным — кодирующая последовательность. Длина такого фрагмента — 4155 п.о. Он кодирует белок, похожий на обонятельный рецептор 1J1. Последовательность данного фрагмента получена следующим образом: сначала был получен скэффолд с интересующим нас идентификатором, потом последовательность фрагмента (который расположен на комплементарной цепи) помещена в файл:
seqret GCF_004126475.2_mPhyDis1.pri.v3_genomic.fna:NW_023397510* -out scaff_seq.fasta
seqret scaff_seq.fasta[26409:30563:r] contig_seq.fasta
Будем работать с полученной последовательностью. Запустим по ней алгоритм blastn, исключив из поиска находки, относящиеся к хордовым. Чтобы увидеть все находки пришлось изменить параметр максимального числа находок до 500. Получился 321 результат, причём все они являются синтетически полученной ДНК. Наверно, этим термином называют праймеры. Если перейти по ссылкам результатов, видно, что подобные конструкции важны для исследования открытых рамок считывания (ORF). Имея такие результаты для поиска по blastn, становится ясно, почему по другим алгоритмам отсутствуют результаты: если есть только синтетические ДНК, то данных для белков не будет.
Но другие алгориты BLAST тоже важны. Каждый из них можно использовать при решении определённой задачи. Например, если нужно сравнить последовательности тРНК группы организмов, то можно воспользоваться blastn. А если необходимо узнать, на какой хромосоме полностью отсеквенированного генома организма расположена последовательность, кодирующая изучаемую тРНК, пригодится megablast. Если есть последовательность, кодирующая мембранный белок бактерий, и нужно проверить, встречается ли этот белок, у архей, следует использовать blastx, который выполнит автоматическую трансляцию последовательности и осуществит поиск в белковой базе данных. Наконец, если есть последовательность, предположительно кодирующая некоторый белок, и небходимо проверить наличие сходной последовательности у архей, можно использовать tblastx, который выполнит поиск по транслированной нуклеотидной базе данных.
Попробуем найти в геноме нашего эукариота гены основных рибосомальных РНК по далёкому гомологу. В нашем случае далёким гомологом будут 16S и 23S рРНК штамма E.coli. 16S рРНК входит в состав малой субъединицы бактериальной рибосомы и участвует в посадке рибосомы на мРНК, а 23S — в состав большой и играет ключевую роль в образовании пептидной связи. То есть эти рРНК неотъемлемые участники трансляции. У эукариот состав несколько другой, к тому же у них 2 типа рибосом: 80S, 70S (находится в митохондриях или пластидах). 18S рРНК эукариот гомологична 16S рРНК прокариот, но напрямую не связывается с мРНК; 28S и 5.8S рРНК считаются гомологами прокариотической 23S рРНК
Итак, для начала проиндексируем последовательность генома нашего эукариота:
makeblastdb -in GCF_004126475.2_mPhyDis1.pri.v3_genomic.fna -dbtype nucl -out discolor_db.fastaДалее будем пользоваться blastn, так как будем искать сходство последовательностей с рибосомальной РНК (перед этим последовательность для 16S рРНК была взята комплементарная и перевёрнута для расположения 5'-3'):
blastn -task blastn -query 16S_rRNA_ecoli.fasta -db discolor_db.fasta -out res_blast_16S.txt
blastn -task blastn -query 23S_rRNA_ecoli.fasta -db discolor_db.fasta -out res_blast_23S.txtДля 23S рРНК находок было больше, чем для 16S; находки пришлись на разные хромосомы Phyllostomus discolor, какие-то оказались в неопределённых скэффолдах. Результаты для 16S рРНК можно увидеть в файле, а для 23S рРНК — здесь.
При выполнении данного задания ожидалось увидеть обширные выровненные участки, так как последовательности, кодирующие рРНК, очень консервативны. Но в полученных файлах всего несколько выравниваний, длина которых не превышает и пары сотен нуклеотидов (при условии, что длины данных 16S и 23S рРНК составляют 1556 и 2932 п.н. соответственно). Однако стоит отметить, что выравнивания каждой последовательности тяготеют к определённым участкам генома. Возможно, такой необычный результат является следствием того, что рРНК закодированы в геноме в виде кластеров с большим числом повторов, а, как известно, повторяющиеся последовательности "доставляют некоторые неудобства" при сборке генома.