HMM-профиль семейства белков и проверка его работы
На сайте Pfam был выбран домен Carbohydrate binding module 77 (ID: CBM77, AC: PF18283), удовлетворяющий выставленным критериям:
- Число последовательностей в full - 96 (больше 40, но меньше пары сотен);
- Средняя длина домена - 108.1 (менее 150);
- Среднее сходство (identity) - 42%;
- Средний процент покрытия последовательности белка доменом (coverage) составляет 11.82% (есть место для второго домена);
- Число доменных архитектур - 22.
Число белков с данным доменом в full, seed, Uniprot (из вкладки "Alignments") составляет 96, 13 и 412 соответственно. Длина HMM-профиля (вкладка "Curation & model") - 109.
Под критерии доменной архитектуры попадают лишь две: первая с Pectate_lyase_4, CBM77-доменами (41 последовательность), вторая - с Pectate_lyase_4, CBM77-доменами, Por_Secre_tail (25 последовательностей). Для работы взяли первую архитектуру, с Pectate lyase (ID: Pectate_lyase_4, AC:PF00544) доменом. Домен CBM77, расположен после домена Pectate_lyase_4 (рис.1).

Скачиваем последовательности нашего семейства (full.fasta). Сохраняем в отдельный файл с помощью python их AC (AC_from_domain.txt), проверяем наличие дубликатов в excel посредством условного форматирования (в итоге, оказались три пары дубликатов: A0A1Y6C4C1_9ALTE, A0A2G5L0T8_9BACT, A0A1I0QZE8_9FIRM), редактируем файл с последовательностями и AC всех белков, оставляя по одной последовательности из дубликата.
АС последовательностей белков внутри одной архитектуры находим на Pfam, копируем, вставляем в промежуточный файл raw_protAC.txt. Нормальный список получаем с помощью python. Программы приведены ниже.
Python Scripts
# download all sequences in domain (1)
with open('full.fasta', 'r') as file, open('AC_from_domain.txt', 'a') as out:
for line in file:
if line.startswith('>'):
line1 = line.strip().split()
print(line1[0][1:], file=out)
else:
continue
# download all proteins` sequences AC that are in domain architecture (2)
with open('raw_protAC.txt', 'r') as file, open('AC_of_proteins.txt', 'a') as out:
for line in file:
line1 = line.strip().split()
print(line1[0], file=out)
# split AC for EMBOSS (3)
with open('AC_of_proteins.txt', 'r') as file, open('AC_for_emboss.txt', 'a') as out:
for line in file:
line1 = line.split('_')
print(line1[0], file=out)
# count sequences (4)
with open('domain.fasta', 'r') as file:
cnt = 0
for line in file:
if line.startswith('>'):
cnt += 1
print(cnt)
# information about length of sequences from full.fasta (5)
with open('AC_from_domain.txt', 'r') as file1, open('len.txt', 'r') as file2, open('length.txt', 'a') as result:
AC = []
length = []
for line1 in file1:
mod_line1 = line1.strip()
AC.append(mod_line1)
for line2 in file2:
mod_line2 = line2.strip()
if mod_line2.isdigit():
length.append(mod_line2)
else:
continue
for i,j in zip(AC,length):
print(i,j, file=result)Далее получим последовательности белков с выбранной архитектурой с помощью bash и EMBOSS, предварительно поработав с АС белков, чтобы EMBOSS не ругался (питоновский скрипт написан выше). Их получилось 39 из 41, потому что две последовательности (A0A4R6MMW4_9BACL, A0A561Q094_9BACL) удалили из Uniprot 19 января 2022 года. Поэтому их скопируем из full.fasta и добавим в файл.
Программы для скачивания последовательностей (1) и информации о длине последовательностей из full.fasta (2) (для удобства работы в excel с помощью скрипта были получены списки АС со соответствующими длинами):
Bash-EMBOSS Scripts
(1) for i in $(cat AC_for_emboss.txt); do seqret -filter "uniprot:$i" | cat >> domain.fasta; done
(2) for i in $(cat AC_domain_emboss.txt); do echo "$i" >> len.txt; infoseq "uniprot:$i" -filter -only -length | cat >> len.txt; doneПостроим выравнивание в Jalview с помощью алгоритма muscle.
Согласно данным Pfam, координаты первого домена (Pectate_lyase_4) - 284:472, а второго (CBM77) - 661:767. Исходя из этого в нашем выравнивании обрезали участки перед первым и после второго домена, затем убрали несколько последовательностей, которые плохо выровнялись и которые имели 99-100% сходство. Fasta-файл с новым выравниванием.
- Построим HMM-профиль нашей двух-доменной архитектуры с помощью hmm2build:
- Откалибруем (нормируем вес):
- Проведём поиск профиля по последовательностям из full.fasta, получив файл, который используем для таблицы:
hmm2build HMM_profile new_aln.fa
hmm2calibrate HMM_profile
hmm2search -E 0.1 --cpu=1 HMM_profile full.fasta > hmm.txt
Результаты
Google-таблица со всеми необходимыми графиками и колонками.
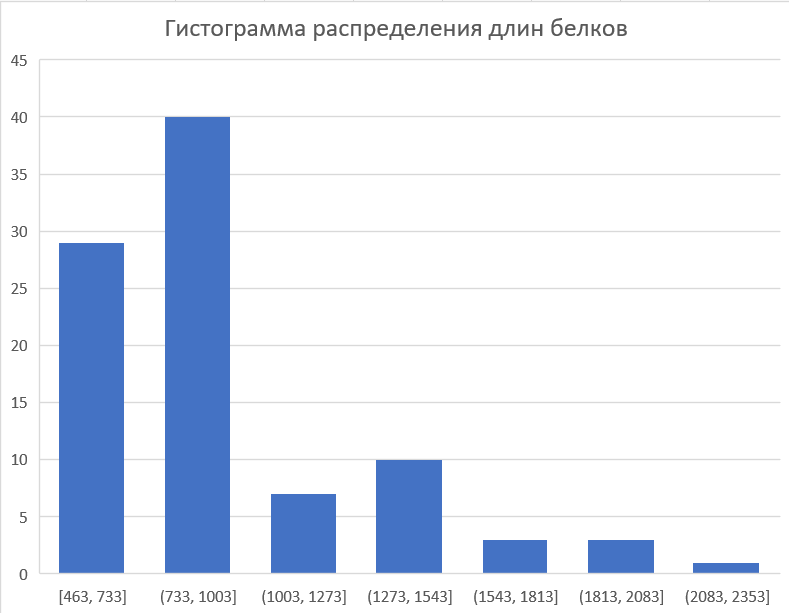
- Видно, что для белков из нашего домена характерна длина в диапазоне 733-1003 (рис.2).
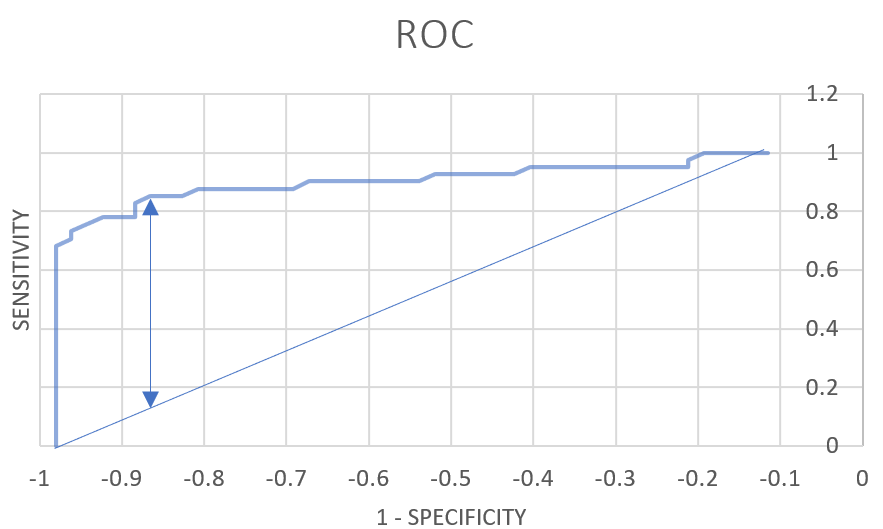
- По RОС-кривой (рис.3) мы можем определить порог: для этого измерим наибольшее расстояние от ROC-кривой до диагонали, соединяющей начало и конец. Здесь эта точка, примерно, с координатами (-0.865384615;0.853658537) и весом 552,90.
- График распределения весов (в google-таблице): график не совсем ровный, потому что примерно после 450 идёт резкий спад до значения ~90.
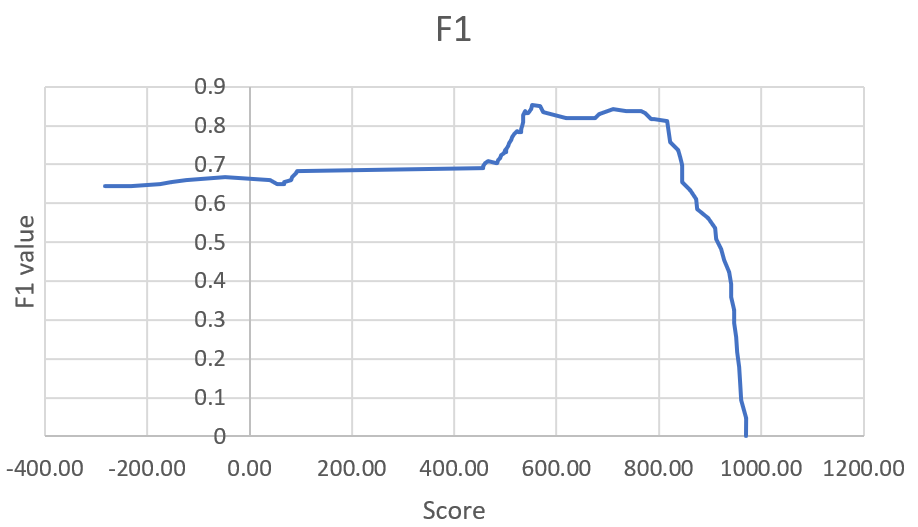
- График F1 (рис.4): при миниальных значениях чувствительности линия плавная, затем растёт, достигает пика и убывает; локальный минимум в точке (618.90;0.820512821).