Практикум 6. ЯМР
Задание 1
В этом задании работали со структурой с PDB-ID 3AKM и 6L7K, которая была получена методом РСА и ЯМР, соответственно. Она представляет собой комплекс белка iFABP из человека и мыши (у 3AKM лиганд - жирная кислота (11-(дансиламино)ундециловая кислота)), меченая флуорофором). Это белок, который связывает жирную кислоту (FABP) и участвует в её транспорте к различным клеточным органеллам, где осуществляется метаболизм, запасание и запуск сигналов. FABPs являются важными регуляторами метаблических и воспалительных процессов, что представляет ценность для разработки таргетной терапии заболеваний, связанных с нарушениями метаболизма[1].
Далее в структуре, полученной методом РСА, выбрали три водородные связи и визуализировали их в PyMol (список и картинки (рис.1-3) приведены ниже). Затем мы построим табличку с расстояниями между донорами и акцепторами в РСА- и ЯМР-модели. Расстояния для РСА-модели измерялись в PyMol, посредством встроенных там инструментов, а для ЯМР - с помощью пакетов BioPython и numpy, чтобы не считать кучу значений вручную (учитывая максимальное, минимальное и среднее значение по всем моделям). Табличку расстояний (в Å) реализуем с помощью модуля pandas (ссылка на код).
- Список водородных связей (ссылка на сессию):
- Между атомами остова в ядре белка: Leu-78 и Tyr-70 (атомы азота и кислорода в пептидной группе). Эти остатки принадлежат двум β-листам.
- Между боковыми цепями в ядре белка: Tyr-14 и Arg-126 (атомы кислорода и азота, соответственно), находящиеся на β-листах.
- В петлях, находящихся на поверхности белковой глобулы: Lys-88 и Glu-107 (атомы азота и кислорода, соответственно), находящиеся на β-листах.
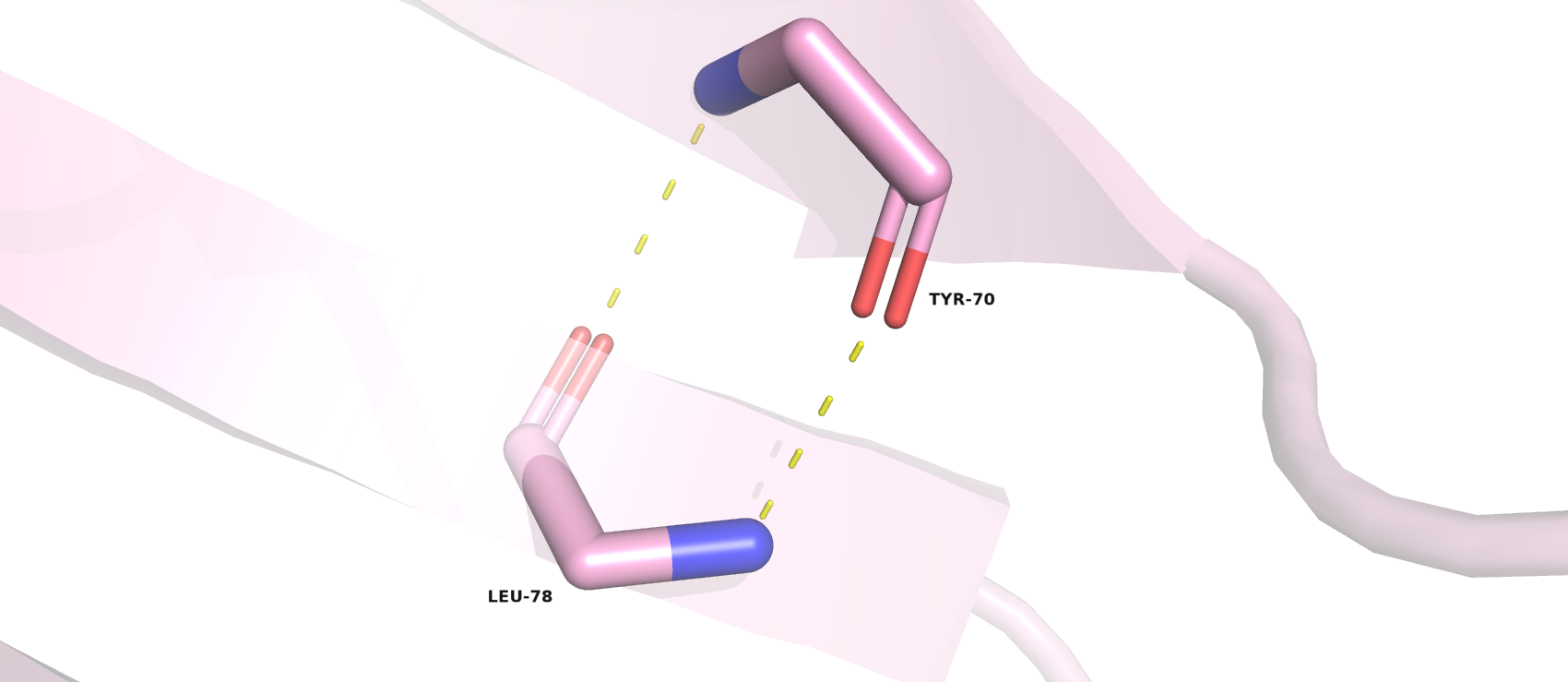
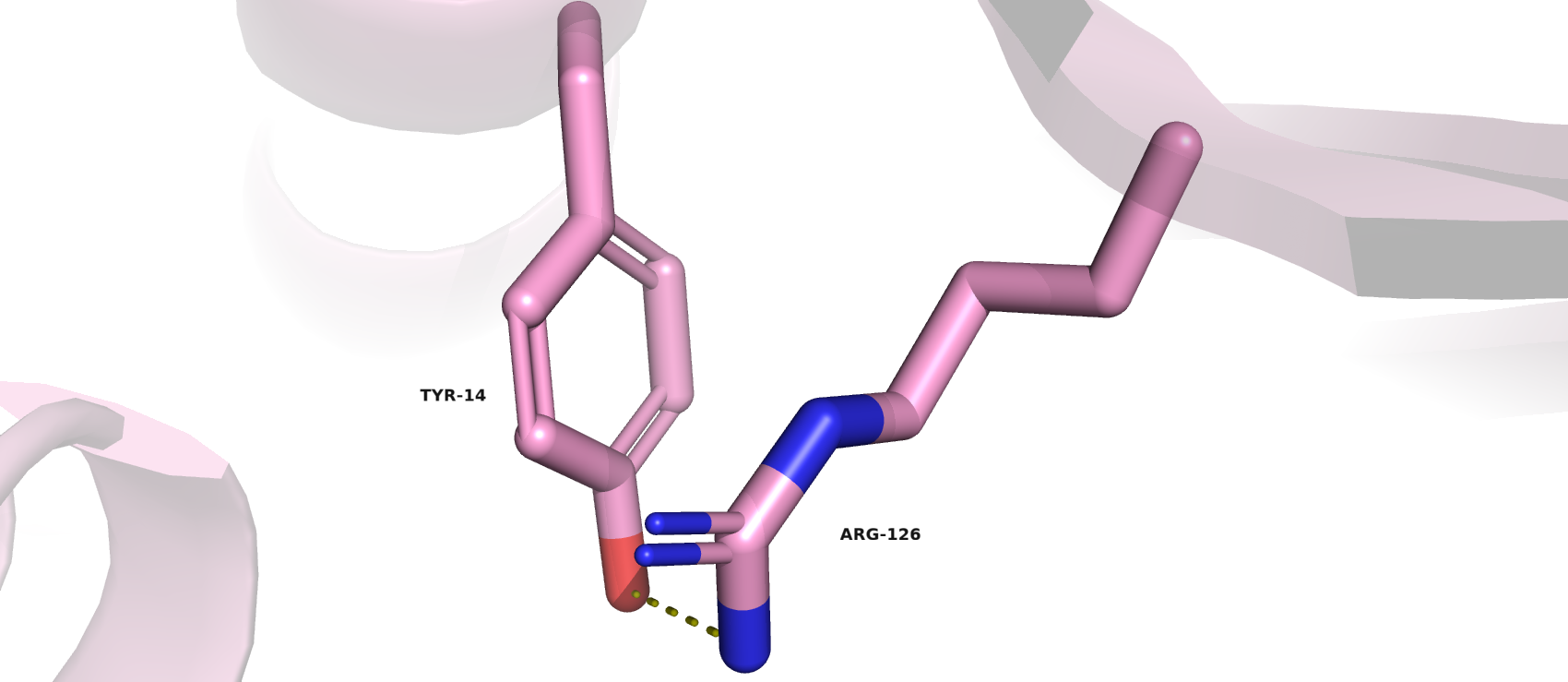
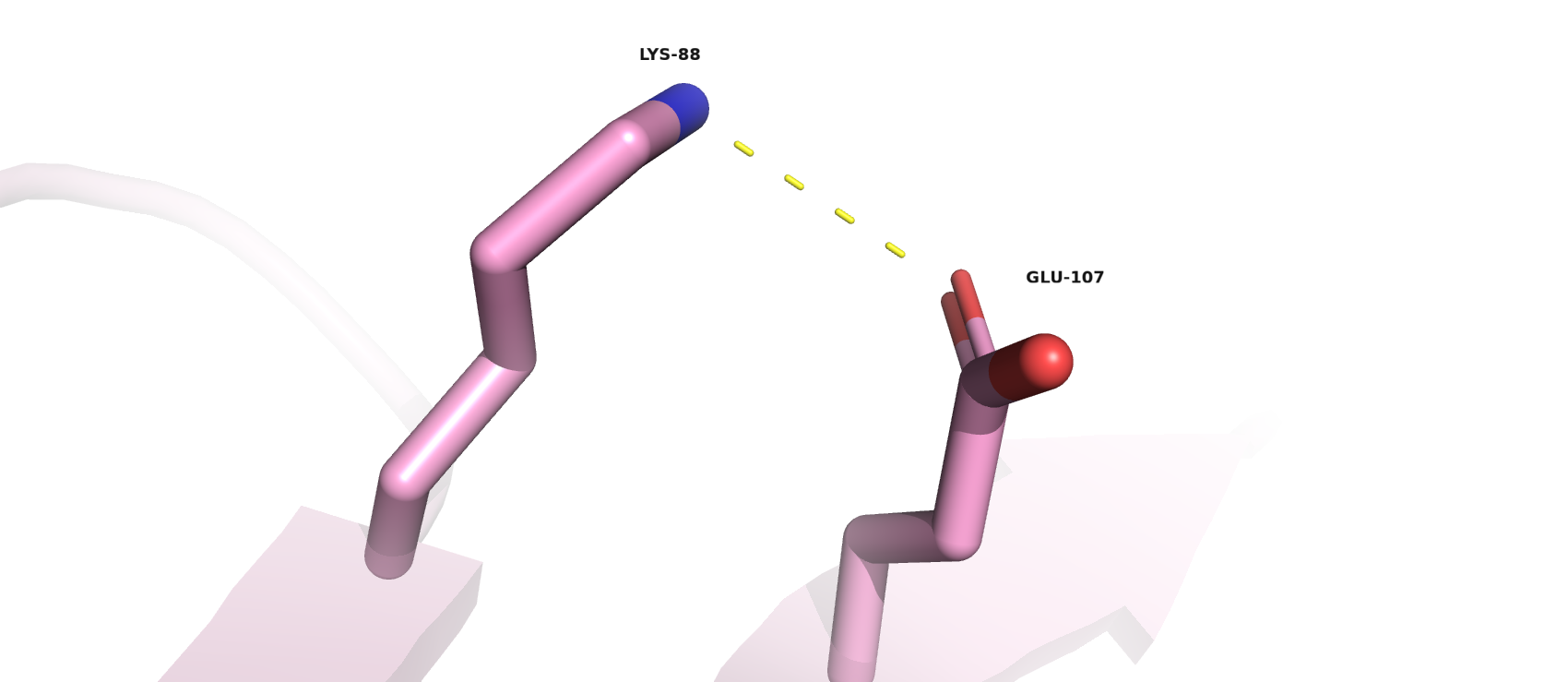
Измерив расстояния между атомами вышеприведённых остатков, обнаружили, что не все связи присутствуют в ЯМР-моделях: либо в определённом проценте моделей, либо ни в одной из них. Скорее всего, это связано с тем, что белок во время ЯМР-эксперимента подвижен, пока в РСА он находится внутри кристалла и, соответственно, неподвижен; возможны также шумы в данных эксперимента, или он не совсем идеален. Поэтому имеет смысл проводить помимо ЯМР-анализа РСА, хотя первый метод оказывается более точным, но не совсем.
Задание 2
До начала работы с Python-пакетом ProDy было проведено выравнивание цепей А и А соответствующих структур. Перед этим были удалены вода и лиганды, поскольку во второй структуре их не было изначально. Структуры выровнились (рис.4), однако появились серые участки в последовательностях, означающие отсутствующие аминокислоты. Дело в том, что они есть в обеих структурах, но в разных конформациях, но при работе с ProDy мы это не учитываем (это не аминокислоты, которых нет в структурах; срезы мы берём по РСА-модели, так как там несколько цепец и лиганды). В jupyter-блокноте представлен код, где вычисляются средние RMSF и B-факторы по всем остаткам и делается вывод о существовании связи между параметрами.
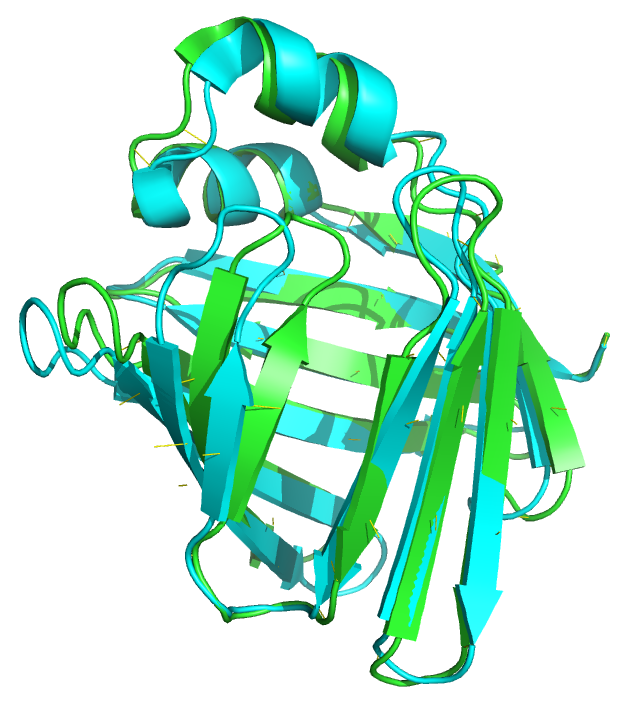
Задание 3
В этом же jupyter-блокноте мы работаем с кодом, но уже для вычисления квадрата RMSF по формуле, представленной в статье[2]. Мы его сравниваем с другими значениями квадрата RMSF, реализованном в пакете ProDy, и делаем вывод на этой основе (написан в jupyter-блокноте).
Список литературы
- Xiao T, Lu Y, Fan JS, Yang D. Ligand Entry into Fatty Acid Binding Protein via Local Unfolding Instead of Gap Widening. Biophys J. 2020 Jan 21;118(2):396-402. doi: 10.1016/j.bpj.2019.12.005. Epub 2019 Dec 14. PMID: 31870540; PMCID: PMC6976794.
- Kuzmanic A, Zagrovic B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys J. 2010 Mar 3;98(5):861-71. doi: 10.1016/j.bpj.2009.11.011. PMID: 20197040; PMCID: PMC2830444.