Вернуться на страницу семестров
D2 (совмещение структур)
Задание 1. Построить совмещение структур белка 1H7M (из задания на ЯМР) и четырех структурных гомологов
✧ Для выбранного ранее белка с PDB-кодом 1H7M были найдены 4 гомолога для пространственного совмещения, а именно: 1w42, 3vi6, 3v7q и 3on1. (цепи А). Ниже ссылка на выравнивание и рисунок 1 с совмещением (кликабельно).
Рисунок 1. Пространственное совмещение 1H7M и его гомологов (1w42, 3vi6, 3v7q и 3on1) для цепи А, построенное средствами JMol в PDBeFold.
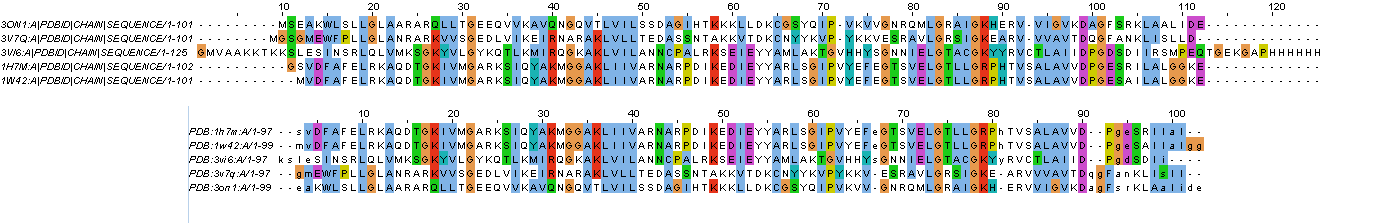
Рисунок 2. Выравнивания: cверху - полученное с помощью сервера ebi muscle, снизу - структурное (в выравнивании по структуре следует смотреть только на большие буквы; маленькие буквы считаются не выровненными). Посторался картинки подогнать друг под друга.
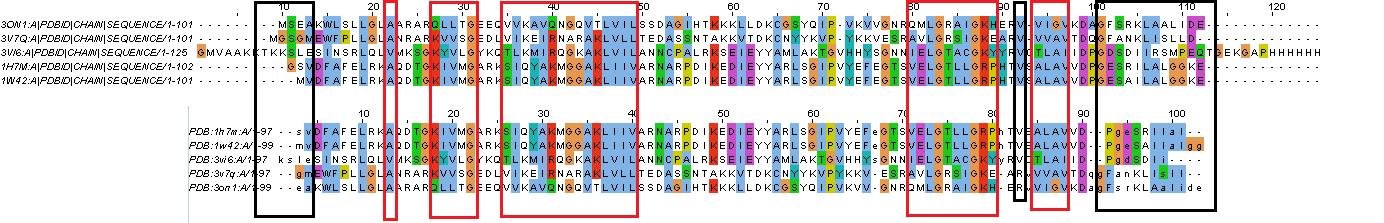
Рисунок 3. Отличия и сходства между выравниваниями: красным показаны сходства в выравниваниях, черным отличия. Видно, что наиболее сходные участки в выравниваниях - элементы вторичной структуры, а отличаются линкеры, связывающие их, а также N- & C-концевые участки. Однако, есть одно интересное исключение, консервативный Val в позиции 92, в выравнивании по структуре у двух гомологов меняется на Arg.
Выравнивание (ebi muscle)
Выравнивание структур (PDBeFold)
Проект в формате jar с двумя выравниваниями
✧ Вывод к заданию 1: можно заметить, что оба выравнивания (и с помощью Muscle, и пространственное) получились хорошими. Это видно, и по выровненым положительно-заряженным аминокислотным остаткам, и по глицинам, которые обычно консервативны; хорошо выравниваются блоки, соответствующие элементам вторичой структуры (альфа-спирали и бета-листы), поскольку они являются более консервативными структурами. Однако, есть места в выравнивании, которые отличаются, это например N- & C-концевые участки, а также петли между элементами вторичной структуры, что видно из рисунка 3.
Задание 2 (дословно): Используйте поиск по сходству структур в PDBeFold (бывш. SSM). Осуществите поиск структурных гомологов для одного из доменов: 1b0m A:203-315, 1adj C:326-419, 1cii A: 284-384, 1dek A: 33-154. Отсортируйте результаты по RMSD. Найден ли сам такой домен в PDB? Если нет, то почему? (Обратите внимание на параметры на заглавной странице PDBeFold.)
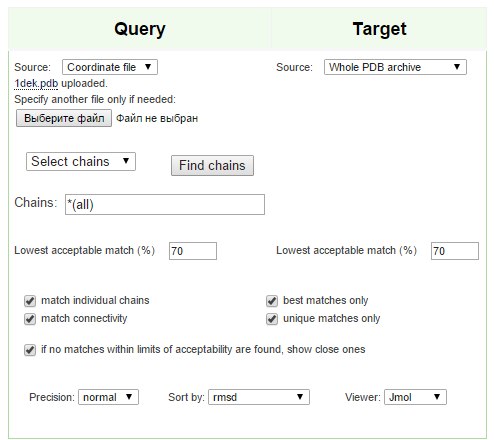
✧ Поскольку сейчас декабрь, выбор был очевиден :) Выбрал домен 1dek A: 33-154 (цепь A). Для этого в PDB-файле были удалены участки цепи А с 1-32 и 155-до конца, и полностью удалена информация о цепи В (поэтому в окно CHAINS можно было ничего не вписывать) Результаты ниже:
Рисунок 3. Запрос в PDBeFold.
Рисунок 4. Выдача в PDBeFold.
✧ Вывод к заданию 2: видно, что есть одна находка (цепь А белка 2l4b) , а исходного домена не наблюдалось. Это можно объяснить, возможно тем, что использовался очень высокий порог в 70%.
Задание 3: cовмещение по заданному выравниванию
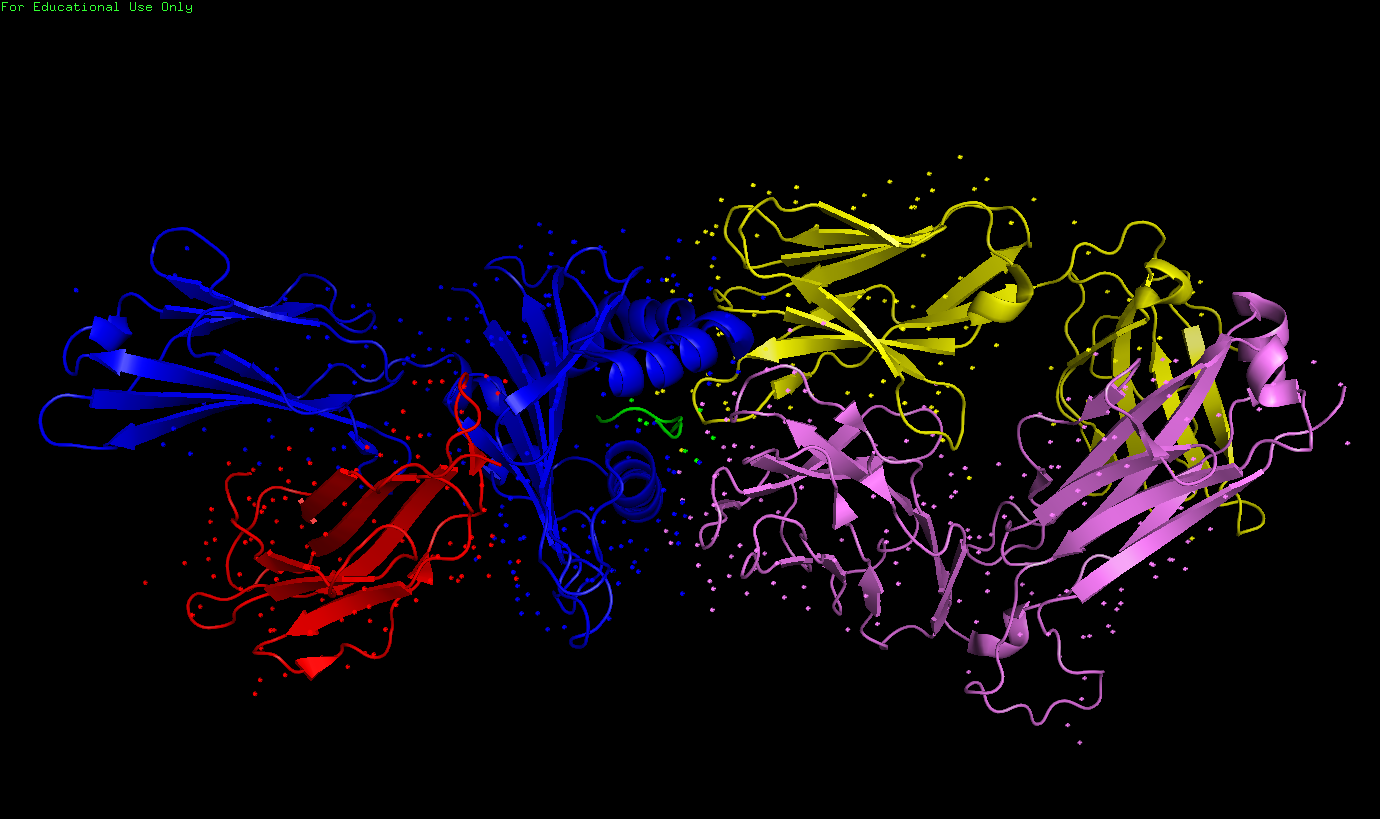
✧ Сохранить в формате PDB одну структуру константного домена T-клеточного рецептора из цепочки альфа и одну из - бета. Выбрал первую выдачу, а именно 1oga (рисунок 5) - region d:118-202 (alpha-chain) и region e:119-245 (beta-chain). Ниже ссылки на PBD-файлы, и изображения.
Рисунок 5. T-клеточный рецептор 1OGA. Показаны все 5 цепей: A - синим, В - красным, С - зеленым, D - желтым и E - фиолетовым. Нас будут интересовать именно D и E. Именно эти PDB-файлы прикреплены ниже, получены командой split_chains
alpha-chain
beta-chain
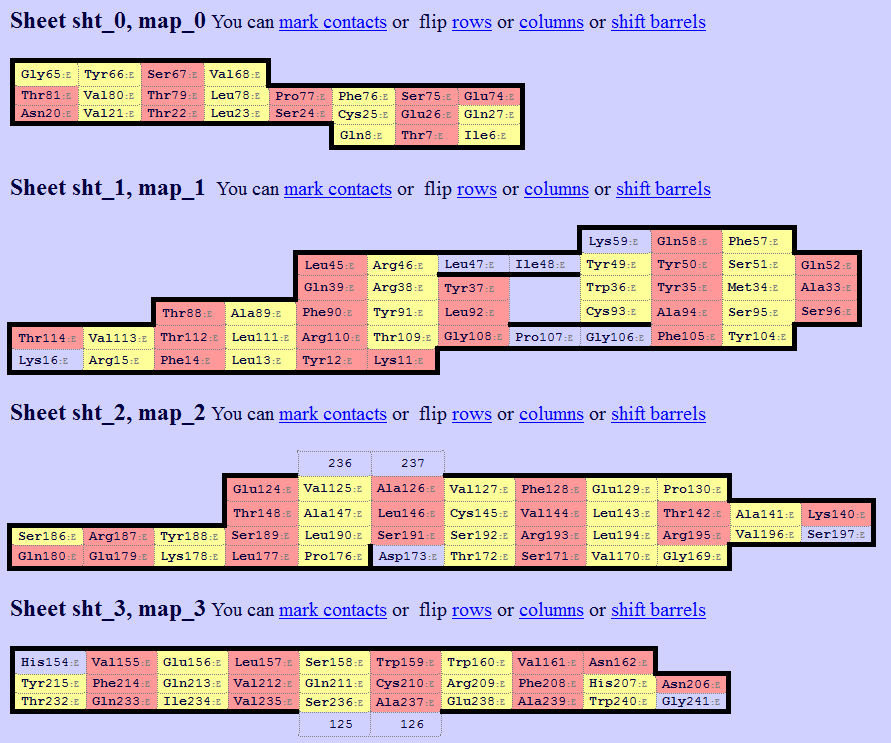
✧ Примечание: я немного модифицировал задание, по нему нужно было выбрать определенные участки доменов, а я сделал задание для полных доменов, так интереснее. Далее пустил эти PDB-файлы на вход программы SheeP (based on DSSP), для того, чтобы получить карты β-листов. Результаты представлены на рисунках 6 и 7. (кликабельно)
Рисунок 6. Выдача SheeP для β-листов α-цепочки
Рисунок 7. Выдача SheeP для β-листов β-цепочки
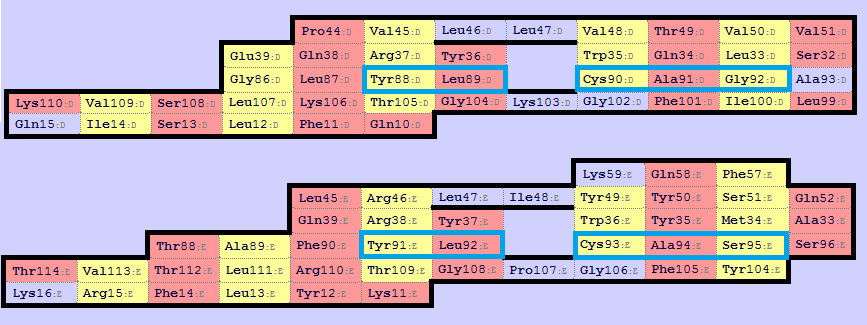
✧ Видно, что второй лист (Sheet sht_1, map_1) α-цепочки соответствует второму листу (Sheet sht_1, map_1) β-цепочки. Так получилось, что карты соответствующих друг другу листов сразу были в одной ориентации, поэтому ничего менять не пришлось. Далее структуры необходимо выровнять по консервативным цистеинам, такими цистеинами являются Cys90 (Sheet sht_1, map_1) α-цепочки и Сys93 (Sheet sht_1, map_1) β-цепочки. Ниже представлено выравнивание с помощью команды pair_fit в PyMol (рисунок 9-10). А на рисунке 8 демонстрируется по каким СА-атомам остаков было проведено совмешение.
Команды, использованные для получения изображения 8.
>
Рисунок 8. Продемонстрировано по каким СА-атомам остаков было проведено совмешение.
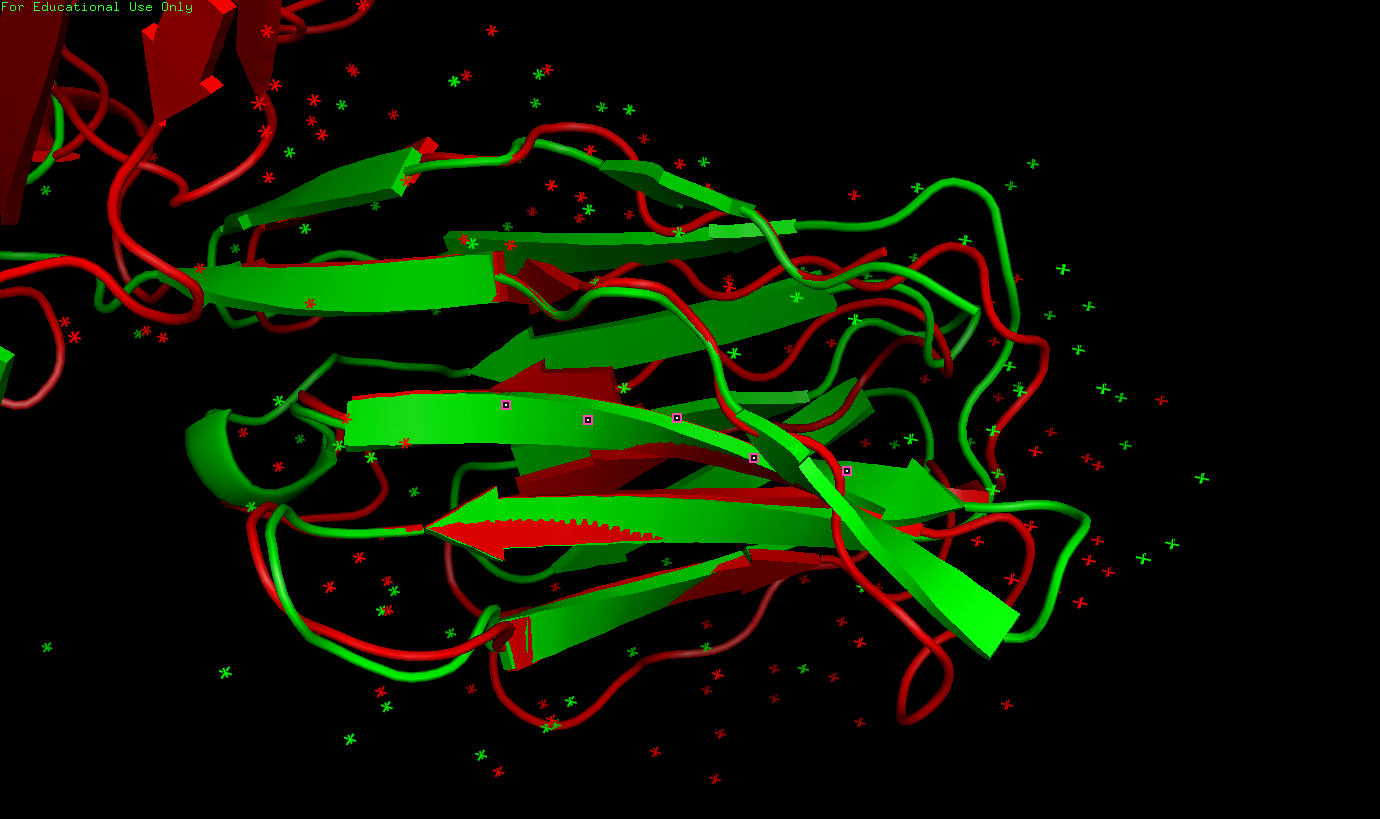
Рисунок 9. Результат команды pair_fit
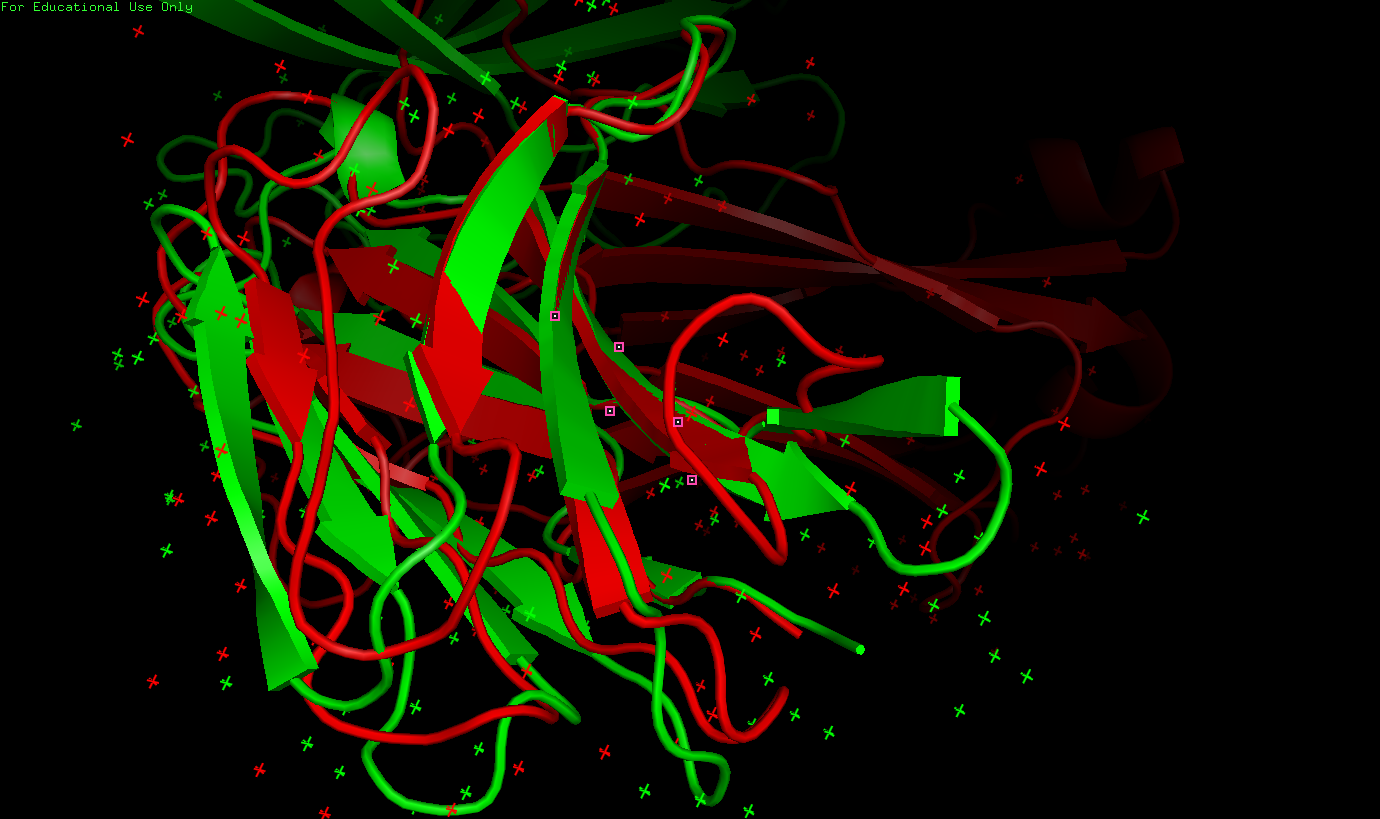
Рисунок 10. Результат команды pair_fit (тоже самое, но с другого ракурса)
✧ Вывод к заданию 3: была проделана большая работа. Показано, что топология для двух цепей не различается (при совпадении топологий каждой петле в одной структуре соответствует петля в другой, что показано на рисунке 9).