Задание 1: разные варианты BLAST для фрагмента ДНК.
Я выбрала участок, находящийся на 5 хромосоме (5q12.1) с координатами 25.536.605-25.560.156 и длиной в 23.552. На этом фрагменте ДНК расположен ген IPO11 (3345 п.о.), кодирующий белок Importin-11, который, в свою очередь, участвует в активном транспорте белков из цитоплазмы в ядро.
Идентификатор нуклеотидной записи: NC_054744.1
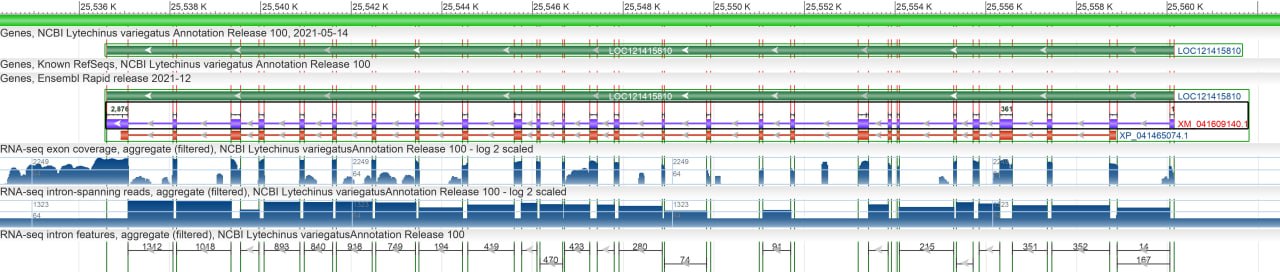
На рис.1 изображен 1 ген, 1 мРНК, 1 CDS, интроны и 27 экзонов.
Длина CDS: 2910 nt.
Ссылка на файл с последовательностью данного участка в FASTA-формате
Поиск BLAST
Целевым таксоном был выбран Cyclostomata- круглоротые из подкласса Черепных, в то время как морской еж является представителем класса Echinoidea. Оба таксона относятся к кладе Deuterostomia (Вторичноротые). По результатам поиска BLAST среди выбранных мною таксонов была построена сравнительная таблица с указанием применяемых программ и параметров поиска.
| Программа | Длина слова | Количество находок |
|---|---|---|
| megablast | 28 | 0 |
| 16 | 2 (плохие значения е-value у находок) | |
| blastn | 11 | 1 |
| 7 | 5 | |
| blastx | 5 | 2 |
| 2 | 2 | |
| tblastx | 3 | 57 (E-value=0.01) |
| 3 | 67 (E-value=0.05) |
Изменение порога значения e-value было сделано для уменьшения количества находок, вероятность попадания в подборку которых высока. Увеличение параметра "wordsize" приводит к уменьшению количества находок и повышает чувствительность поиска. Измененять количество выдаваемых находок не потребовалось, так как во всех случаях их было менее 100.
Алгоритм BLASTN подойдет для поиска схожих нуклеотидных последовательностей с целью определения таксономии, но не следует использовать для поиска гомологов кодирующих последовательностей. Для более чуствительного сравнения дивергентных последоваетльностей лучше подойдет алгоритм TBLASTX, который может найти сходство среди далеких последовательностей, так как мутирование нуклеотидной последовательности происходит быстрее, чем аминокислотной. Но ипользование алгоритма TBLASTX бесполезно для некодирующих РНК и консервативных некодирующих элементов. Также для поиска среди очень похожих последовательностей возможно использование программы megablast. Если известно, что последовательность содержит определенный ген, то для определения его положения в нуклеотидной последовательности можно использовать BLASTX.
Поиск генов основных рибосомальных РНК по далекому гомологу.
Для решения поставленной задачи подойдут программы blastn megablast, так как их применение возможно для некодирующих последовательностей, коими являются гены рРНК, но использование megablast нецелесообразно для поиска по далекому гомологу.
...Индексирование генома Lytechinus variegatus
C:\Program Files\NCBI\blast-2.15.0+\bin добавлен в PATH, затем:
makeblastdb -in "GCF_018143015.1_Lvar_3.0_genomic.fna" -dbtype nucl
Вывод 8 файлов:
GCF_018143015.1_Lvar_3.0_genomic.fna.ndb
GCF_018143015.1_Lvar_3.0_genomic.fna.nhr
GCF_018143015.1_Lvar_3.0_genomic.fna.nin
GCF_018143015.1_Lvar_3.0_genomic.fna.njs
GCF_018143015.1_Lvar_3.0_genomic.fna.not
GCF_018143015.1_Lvar_3.0_genomic.fna.nsq
GCF_018143015.1_Lvar_3.0_genomic.fna.ntf
GCF_018143015.1_Lvar_3.0_genomic.fna.nto
>CP014225.1:complement(926804-928359)|16S_rRNA|Escherichia coli str. K-12 substr. MG1655 strain K-12 >CP014225.1:2234710-2237641|23S_rRNA|Escherichia coli str. K-12 substr. MG1655 strain K-12Эти последовательности, для которых ищутся гомологи, являются последовательностями рРНК рибосом (бактериальных) с коэффициентами седментации 16 и 23 соответственно. 16S rRNA образует 30S субъединицу рибосомы, где она узнает последовательность Шайна—Дальгарно, а 23S rRNA является частью 50S субъединицы. Последовательности 16S tRNA и 23S tRNA почти всегда находятся в одном первичном транскрипте, но в этом случае они удалены в геноме друг от друга и находятся на разных комплементарных одноцепочечных ДНК, вероятно присутствуют две разные копии соответствующих общих последовательностей.
Для быстроты обработки последовательности были вынесены в отдельные файлы. Использовался BLASTN с аргументом blastn, т.к. он больше подходит для отдельных коротких последовательностей и при межвидовом сравнении.
blastn -task blastn -query "16S_rRNA_ecoli.txt" -db "GCF_018143015.1_Lvar_3.0_genomic.fna" -out "16S_rRNA_alignment" -outfmt 7 -evalue 0.1 -word_size 5
Аналогичный запрос для 23S rRNA.Были выбраны весьма мягкие критерии e-value = 0.1 и word size = 7. Принадлежность участков выравнивания (features) была найдена повторным BLASTом онлайн на сайте NCBI с теми же параметрами и результатами.
Было найдено 21 совпадение для 16S rRNA. Все они относились лишь к двум фрагментам последовательности: 887-998 (в одном случае 887-975) и 1494-1536. Выделяется три участка в геноме Lytechinus variegatus, в которых эти выравнивания сконцентрированы, два из них относятся к малой субъединице рРНК эукариот и содержат почти все выравнивания, а один, с единственным выравниванием, относится к неописанному белку.
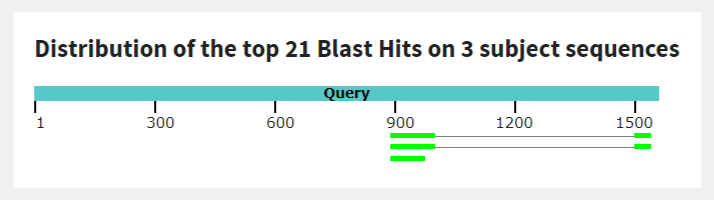
51 выравнивание было найдено для 23S rRNA. Они так же относились к трем участкам в геноме: два из них соответствовали большой субъединице рибосомы, а один не был аннотирован и содержал всего одно выравнивание. На одном из участков, относящихся к большой субъединице рибосомы, было три выравнивания в неизвестном белке, богатом пролином, но это может быть артефактом аннотирования (например ложная рамка считывания), т.к. эти участки совпадают с остальными. В исходной прокариотной последовательности выделяется 5 постоянно совпадающих участков.
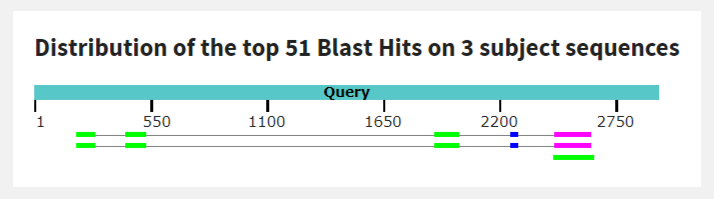
Таким образом 20 из 21 и 50 из 51 выравниваний по рРНК, вероятно, относятся к различным копиям родственных рРНК эукариот в геноме Lytechinus variegatus. Исходя из повторяющихся выравниваний одинаковых отрезков искомых бактериальных тРНК можно предположить 10 копий тРНК большой субъединицы 10 копий тРНК малой субъединицы в геноме Lytechinus variegatus.
На самом деле эти выравнивания можно рассматривать как 10 и 10 выравниваний с большими гэпами, по два консервативных фрагмента от 16S tRNA и по пять от 23S tRNA в каждом выравнивании соответственно, т.к. фрагменты имеют одинаковую принадлежность и расстояние друг от друга. Таким образом на основании данных, полученных BLAST, можно выделить в сумме семь особо консервативных участков тРНК, сохраняющих от 68 до 88% последовательности как у эукариот, так и у прокариот. Возможно, при добавлении в выравнивание 5S rRNA таких участков было бы больше.