Практикум 11
Автор старался, но не может гарантировать отсутствие биологических ошибок.
Домены и профили
Несколько слов про домен и подсемейство.
Еще при заполнении формы в классе был выбран домен Sialidase, N-terminal domain
(PF02973).
Он встречается у O-гликозилгидролаз, сиалидазы гидролизуют гликозидные связи концевых сиаловых остатков
(остатки полиоксиаминокислот)
в олигосахаридах, гликопротеинах, гликолипидах, коломиновой кислоте и синтетических субстратах,
по некоторым данным,
могут выступать в качестве патогенных факторов при микробных инфекциях.
Выделяют 74 архитектуры, содержащих данный домен, была выбрана
PF02973
- PF13088,
как двухдоменная и содержащая оптимальное количество представителей -
126
белков. Второй домен: BNR repeat-like domain.
Подготовка
Были скачены последовательности подсемейства, выровнены в Jalview (Mafft with defaults). Для проверки того, что выравнивание на участках обоих доменов было местами удовлетворительным, отсортировали выравнивание в Jalview по id. Для первых нескольких последовательностей координаты начала и конца доменов согласно pfam:
- 2-166; 397-572 (A0A098ZPR4)
- 80-271; 499-726 (A0A0H2ZQ85)
- 44-223; 457-660 (A0A0J0B100)


Осталось только провести ревизию вырванивания.
Обнаружены и удалены были две последовательности с крупными деляциями в районе доменов.
Из кластеров высокосходных последовательностей оставили одну - порог remove redundancy постепенно снижали
со 100% и в итоге довели до одного представителя в класетере из идентичных на 94% и более последовательностей.
Остались 60 последовательностей.
Материал для построения профиля - выравнивание после ревизии.
В качестве материала для положительного контроля решено взять все последовательности подсемейства.
Материал для калибровки профиля
- два подсемейства семейства с Sialidase доменом,
не пересекающееся с выбранным подсемейством:
PF02973 - PF13385 - PF13088 - PF00754 - PF07554 - PF07554 - PF07554 - PF07554 (6 белков)
и PF02973 - PF13859 - PF00963 - PF00404 (6 белков),
а также для дальнейшей работы необходимы последовательности белков изучаемого подсемейства.
Создание HMM-профиля подсемейства PF02973 - PF13088
Для построения HMM-профиля были выполнены представленные ниже команды.
hmm2build hmmout reviewed.fasta
hmm2calibrate hmmout
hmm2search --cpu=1 hmmout negative.fasta > results.txt
hmm2build делает профиль по выравниванию - результат. Далее после калибровки статистик поиска, проводили сам поиск по материалам для положительного контроля - результат.
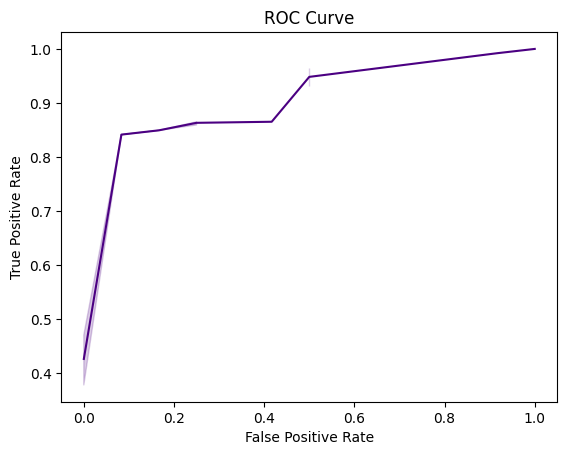
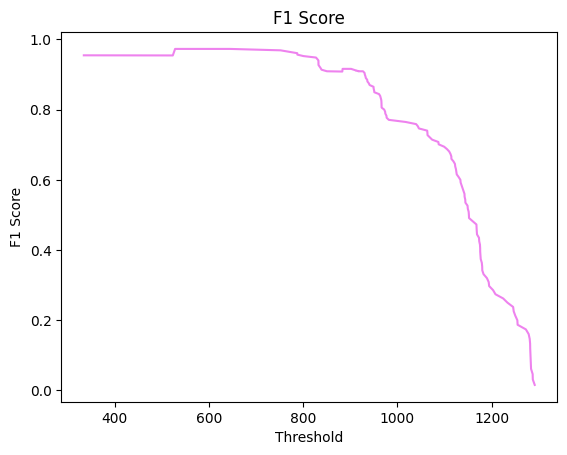
Анализ HMM-профиля
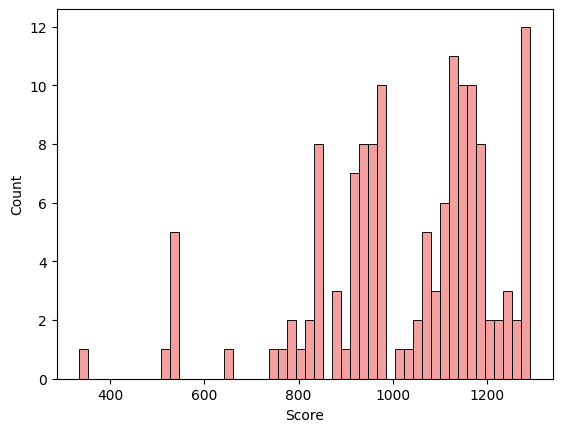
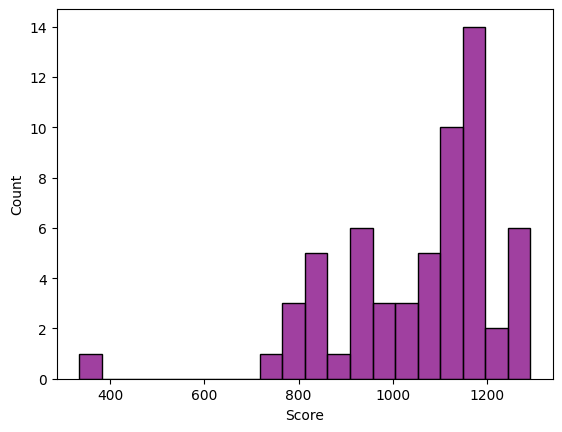
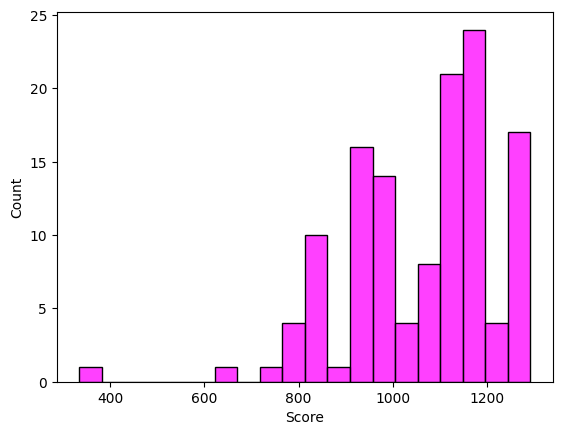
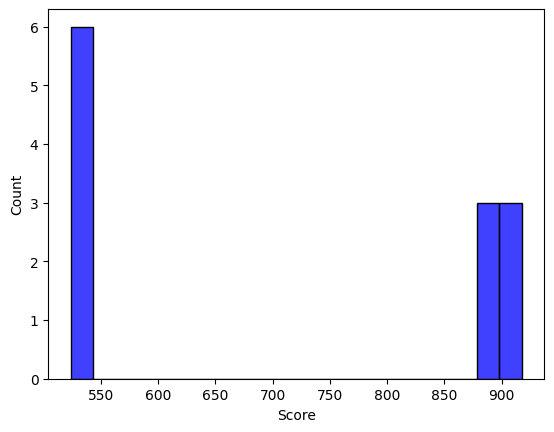
Итоговая таблица, гистограммы весов и графики были получены при помощи скрипта на питоне - colab. Ссылка на таблицу. Гистограммы представлены на рисунках 3-6. Уже из 3 рисунка видно, что у нас отсуствуют отрицательные веса, что уже вызывает подозрения. В рисунке 5 становится видно, что представители негативной выборки плохо выделяются полученной моделью. Графики подтверждают неточное восприятие нашей моделью архитектуры. По гистограммам и графикам видно, что порог либо в 550, в 900 можно брать, наверное, лучше 900.