Домашнее задание 1. Электронная плотность
Автор не был готов к тому, что снова будут нужны kodomo и pymol🥺
Задание 1. ЭП: хорошая и плохая расшифровки
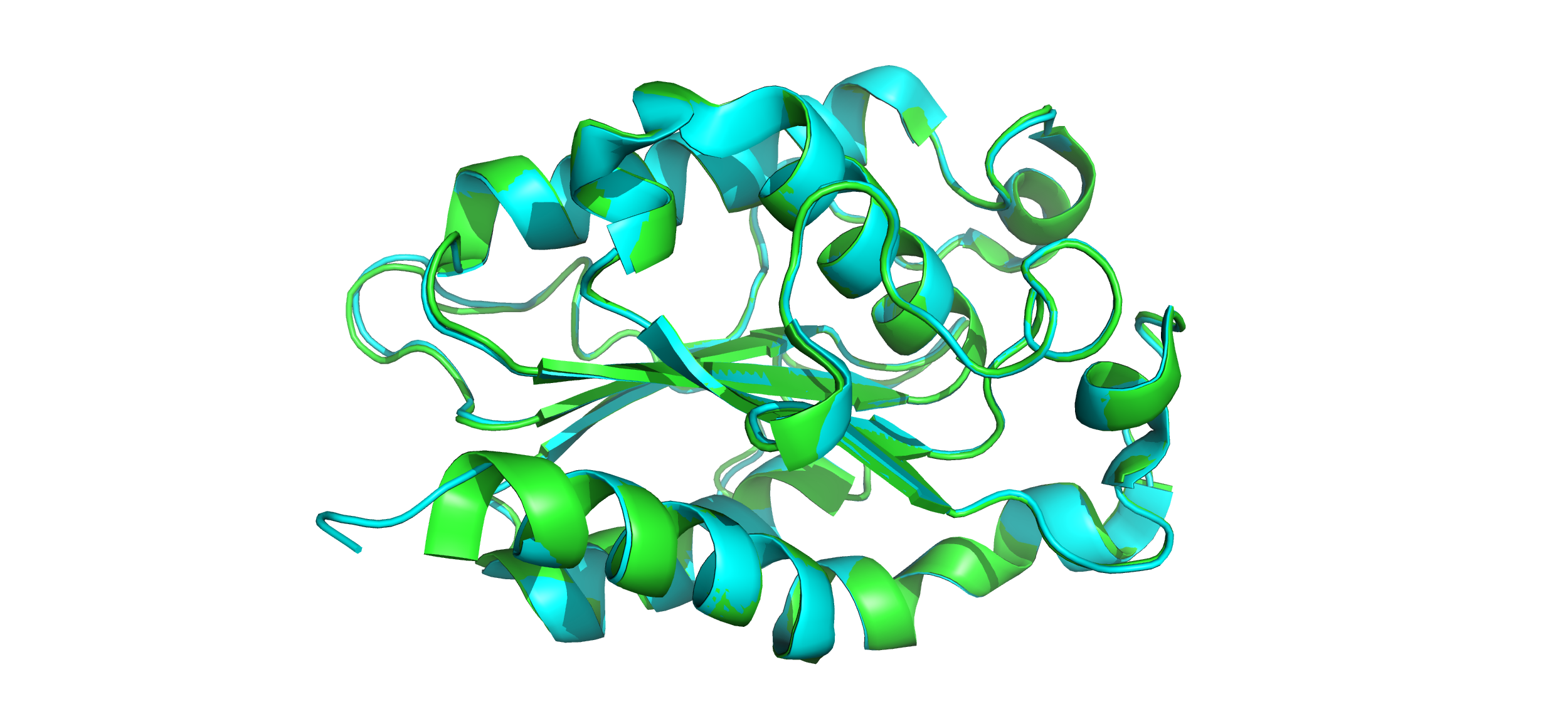
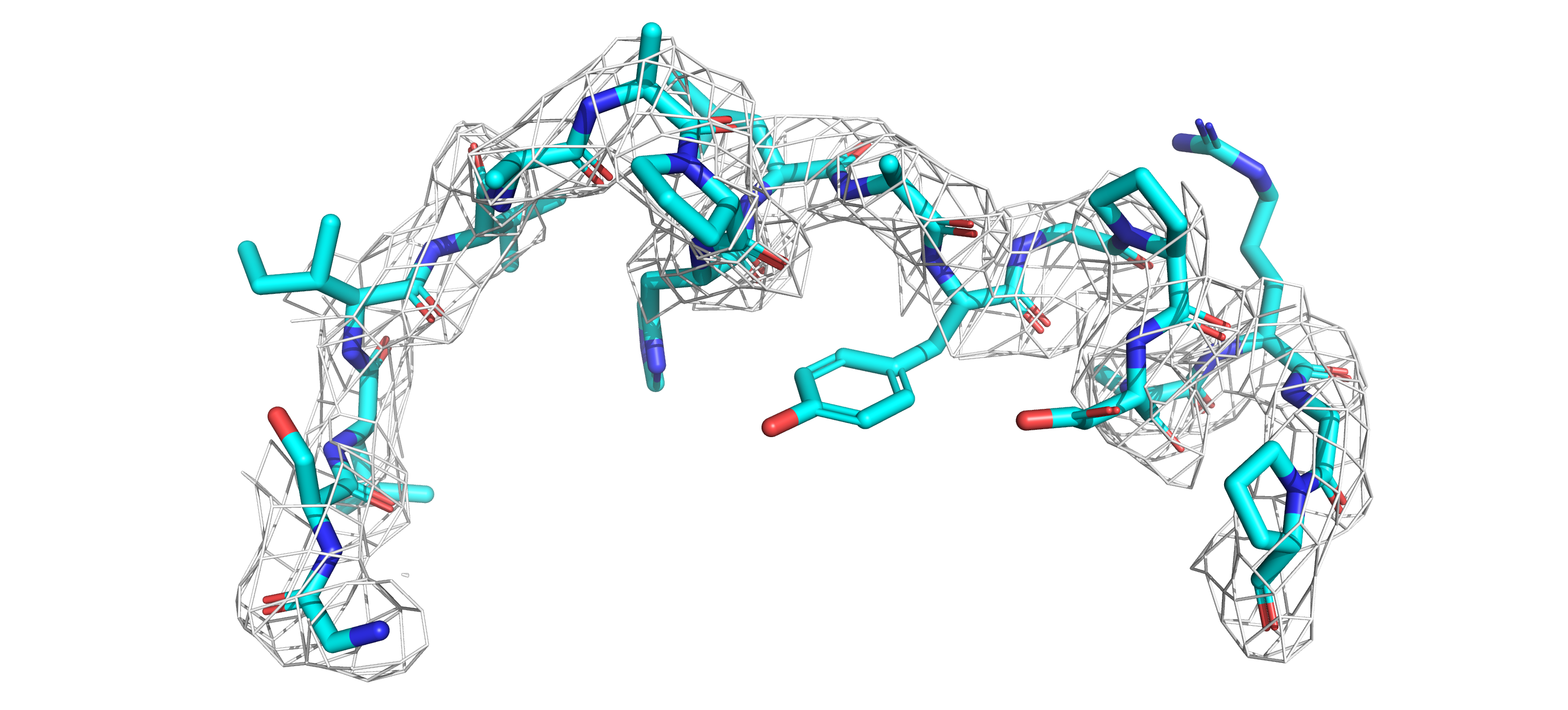
Мне были даны две модели 3QPA и 3ESB. Это кутиназа (относится к классу гидролаз) из Fusarium vanettenii. Выравняла модели при помощи команды (рисунок 1):
super 3QPA, 3ESB
Значимых отличий, которые сразу бросаются в глаза, мне не удалось выявить. Остовы двух белков в целом идут
сходным образом, сборки явно отличаются только на N- и C- концах белков (C-конец можно увидеть в левой нижней
части рисунка 1).
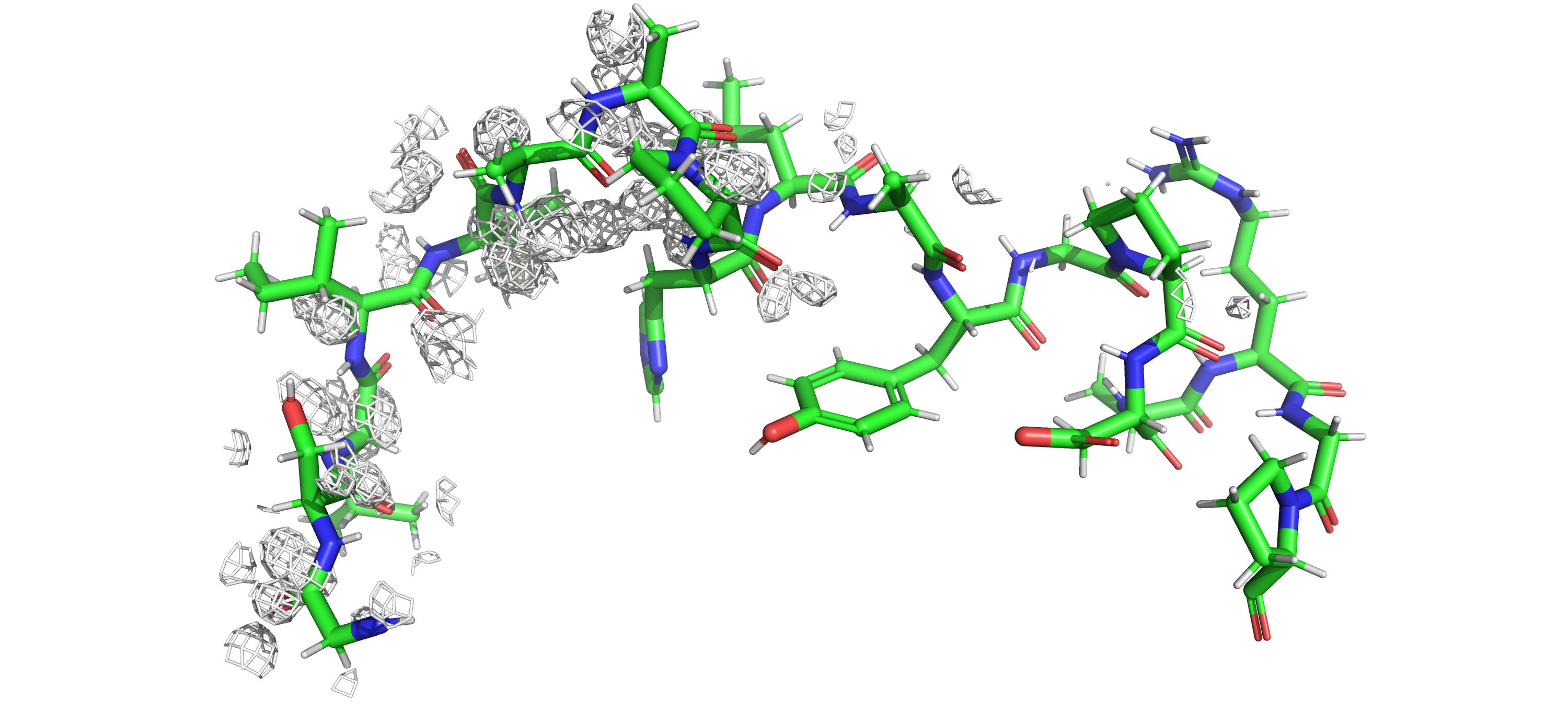
Изучим карты электронных плотностей с помощью mesh. Изначально было решено отображать следующий участок:
195-198 остатки (Ala-Arg-Gly-Pro) - часть одной из альфа-спиралей. Здесь и далее будет показана электронная плотность
вокруг остова и при carve = 2 (если не оговорено иначе), при ином значении ЭП вокруг остова начинала теряться.
Карты ЭП на уровнях подрезки 1 и 2 для участка 195-198 модели 3QPA показали... ничего (рисунок 2):
isomesh mQPA, 3QPA_2fofc, 1, (seleQPA), carve=1 isomesh m2QPA, 3QPA_2fofc, 2, (seleQPA), carve=2
Для модели ESB уже можно наблюдать как меняется отображение ЭП на разных уровнях подрезки (рисунки 3 и 4):
isomesh mESB, 3ESB_2fofc, 1, (seleESB), carve=2 isomesh m2ESB, 3ESB_2fofc, 2, (seleESB), carve=2
Видно, что на уровне подрезки 2 электронная плотность сосредотачивается ближе к остову белка. При этом ЭП идет каркасом вокруг всего остова, не концентрируясь в положениях отдельных молекул.
Отсутствие каких-либо ЭП на маленьком участке остова белка привели к тому, что
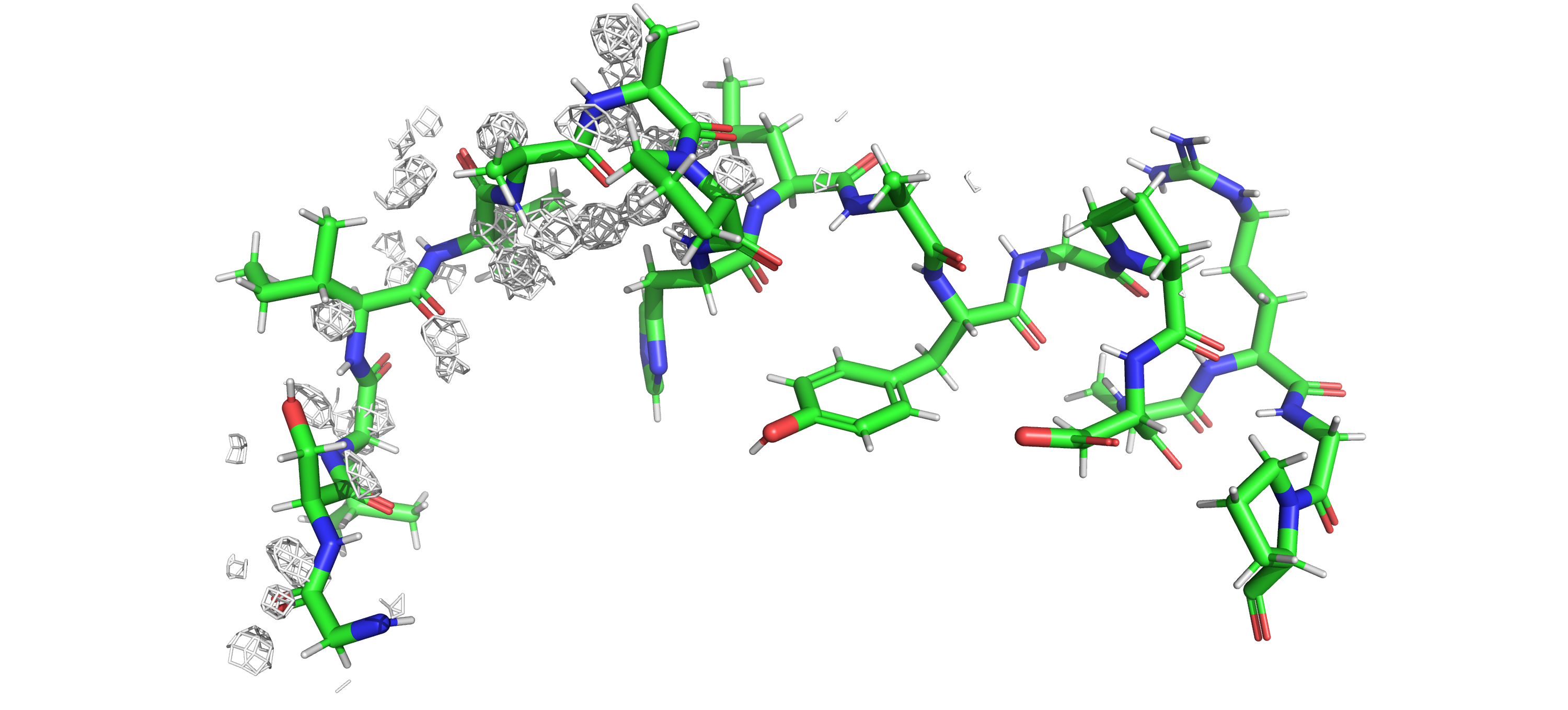
я решила исследовать участок 180-198
для каждой из моделей на уровнях подрезок 1 и 2 (рисунки 5-8).
Для модели 3QPA наконец-то удалось увидеть ЭП на ненулевых уровнях подрезки. ЭП редкая, но значительно
более детализированная, чем у другой модели.
У 3ESB облака ЭП повторяют структуру остова в достаточно общем виде (но зато стабильно!).
Все-таки хоть какие-то облака ЭП мне захоьелось визуализировать для участка 195-198 для модели 3QPA, поэтому я посмотрела на обе на уровне подрезки 0 (рисунки 9-11):
isomesh m0QPA, 3QPA_2fofc, 0, (seleQPA), carve=1 isomesh m0QPA, 3QPA_2fofc, 0, (seleQPA), carve=2 isomesh m0ESB, 3ESB_2fofc, 0, (seleESB), carve=1
Тут стала видна ЭП для модели 3QPA: четко детализованная и сохраняющаяся при понижении carve. ЭП 3ESB же исчезала при понижении значения carve и в целом значительно более размытая и с меньшей атомарной разрешенностью.
Мои визуальные оценки подтвердились данными о разрешении с сайта PDB.
Структура 3QPA с высоким разрешением 0.85Å демонстрирует характеристики,
соответствующие атомарному уровню детализации.
Несмотря на то, что для малого участка 195-198 остатков электронная плотность не была видна
на ненулевом уровне подрезки, при расширении анализа на регион 180-198 или использовании уровня подрезки 0
стала очевидной превосходная детализация плотности - четкая, атомарно разрешенная,
сохраняющаяся при изменении параметра carve.
Структура 3ESB с разрешением 2.30Å показывает типичные для среднего разрешения характеристики:
электронная плотность более размытая, менее детализированная,
но при этом стабильно присутствующая на разных уровнях подрезки.
Однако она не достигает атомарной разрешенности и демонстрирует высокую чувствительность к параметру carve.
Задание 2. ЭП и положение в структуре
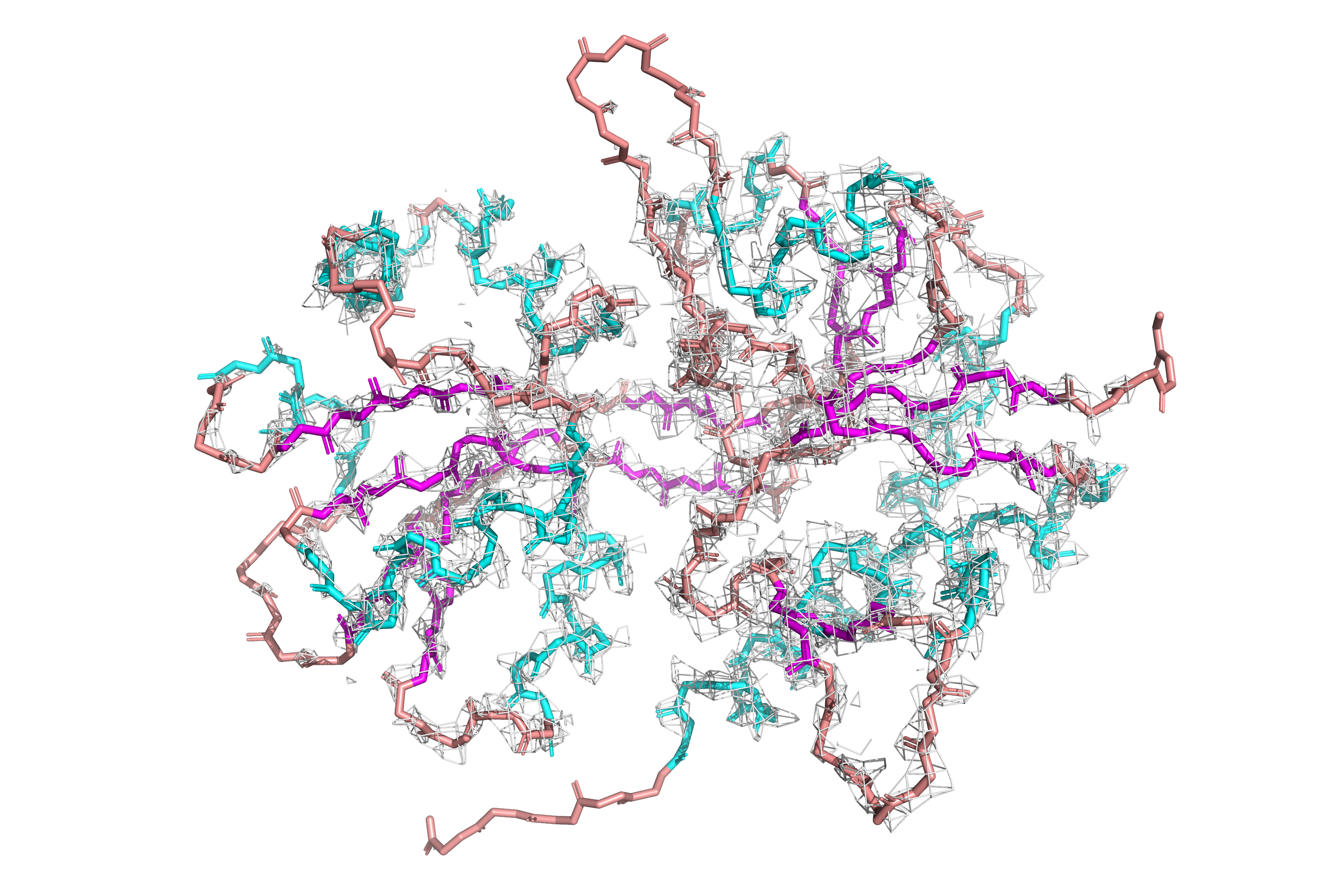
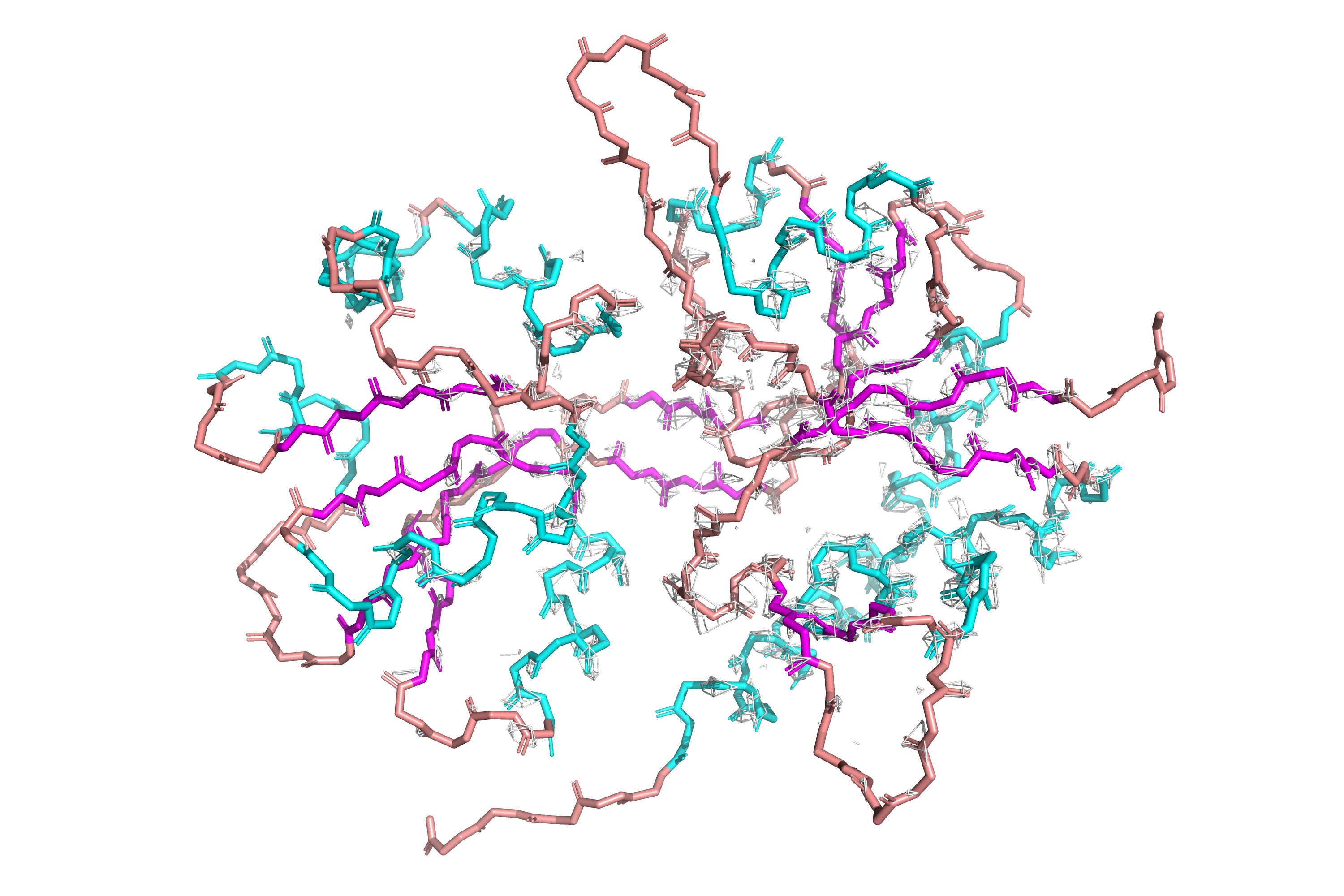
На этот раз работаю с моделью 4G8N домена рецептора GluK3. Общий вид белка представлен на рисунке 12.
Получим структуру и постром карту mesh'ей вокруг остова всего белка на разных уровнях подрезки (значение carve=2 оптимально, при carve=1 ЭП почти отсутствовала):
fetch 4G8N fetch 4G8N, type=2fofc select bb, backbone isomesh m1, 4G8N_2fofc, 1, bb, carve=2 isomesh m2, 4G8N_2fofc, 2, bb, carve=2 isomesh m3, 4G8N_2fofc, 3, bb, carve=2
Карты представлены на рисунках 13-15. Видно, что с повышением уровня подрезки какие-то регионы остова структуры перестают быть покрытыми mesh'ем.
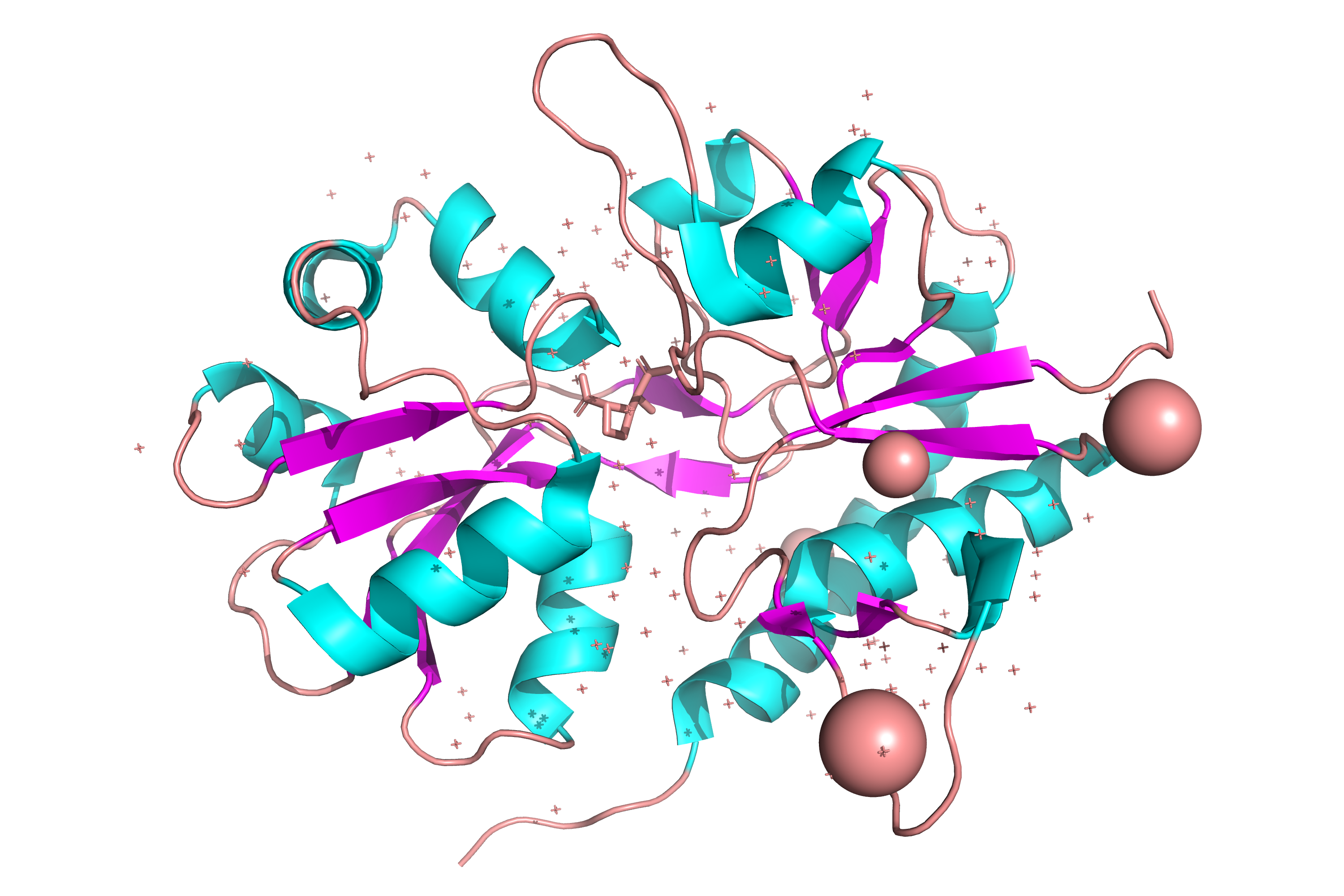
На уровне 1 карта ЭП покрывала практически весь остов белка. На 2 уровне подрезки покрытие начало пропадать
на концах полипептидной цепи, а также на некоторых участках без вторичной структуры. На 3 уровне подрезки
ЭП не покрывала уже участки на границе белка и концентрировалась на отдельных белках (повышение carve приводило
лишь к появлению стороннего шума). Стоит заметить, что на 3 уровне, ЭП если и сохранялась, то
зачастую на карбонильной группе.
Таким образом, с повышением уровня подрезки сохранялась визуализация участков с наиболее отличающей от шума
ЭП. Это более устойчивые участки белка со вторичной структурой и расположенные в наиболее плотно упакованных
частях белка.

Задание 3. Альтернативные положения
Мне был дан вариант задания B: остаток 114-ый Glu на цепи B модели
6RT3. Но что-то в этом не соответствует реальности, поэтому
берем запасной вариант задания B: уже 187-ой остаток Glu на цепи A модели
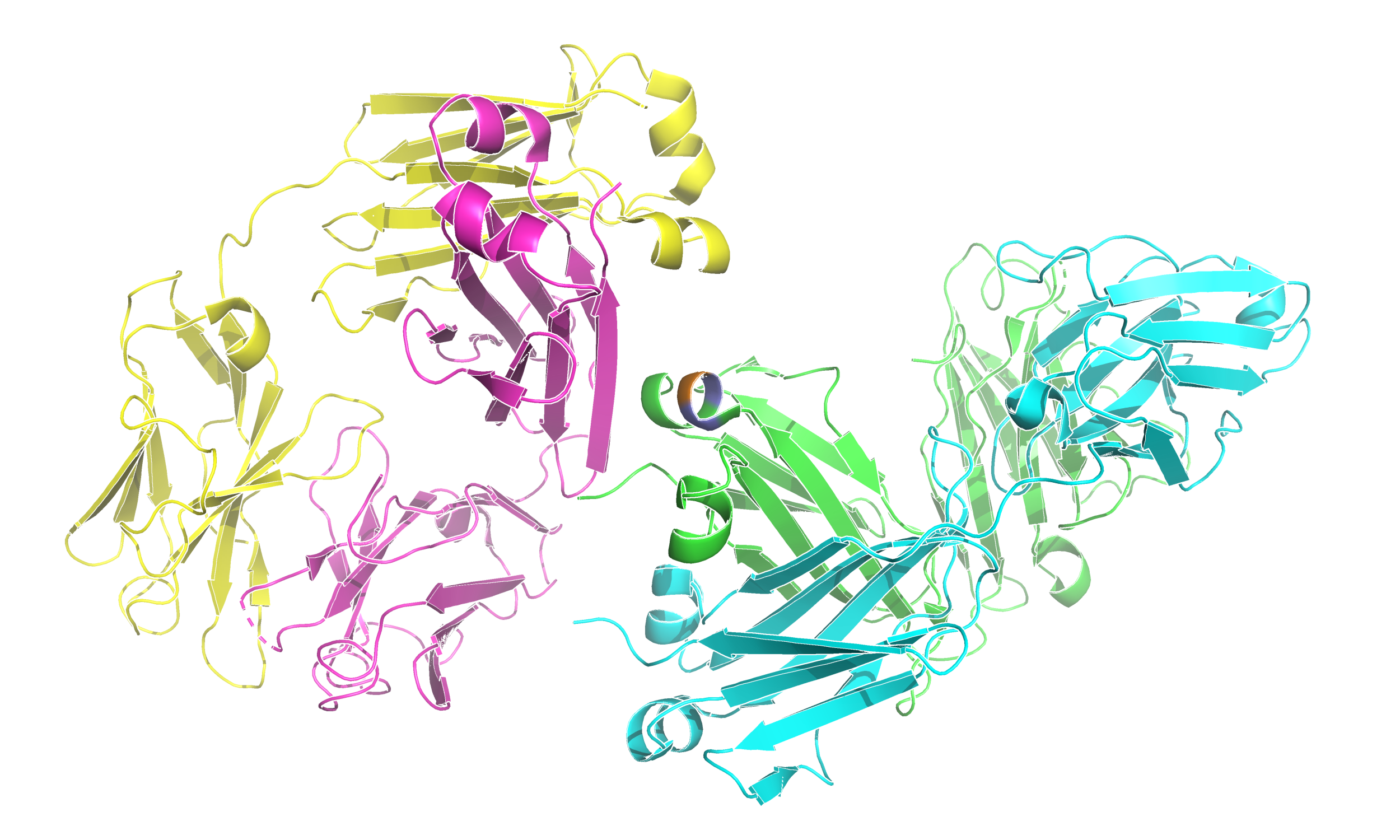
8FO4. Это легкая цепь иммуноглобулина H9 в комплексе еще с чем-то.
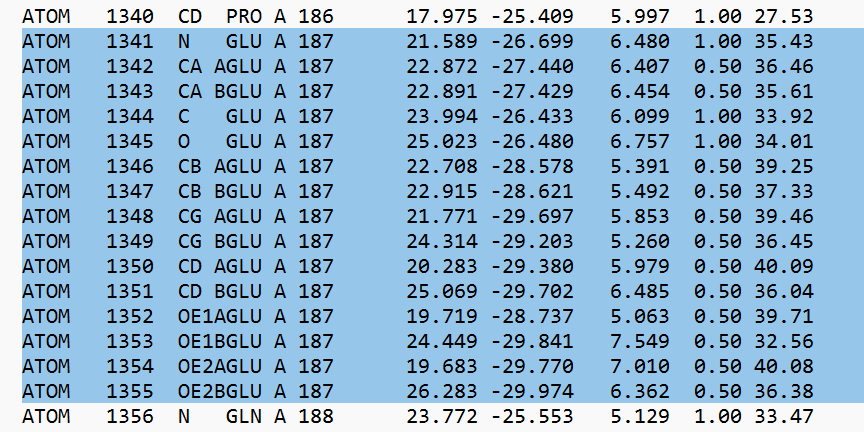
На рисунке 16 показан общий вид белка. Для остатка Glu есть два альтлока с одинаковым occupancy: 0.5 (рисунок 17).
Визуализируем альтернативные конформации глутамата:
fetch 8FO4 select my_residue_A, resi 187 and alt A and chain A select my_residue_B, resi 187 and alt B and chain A
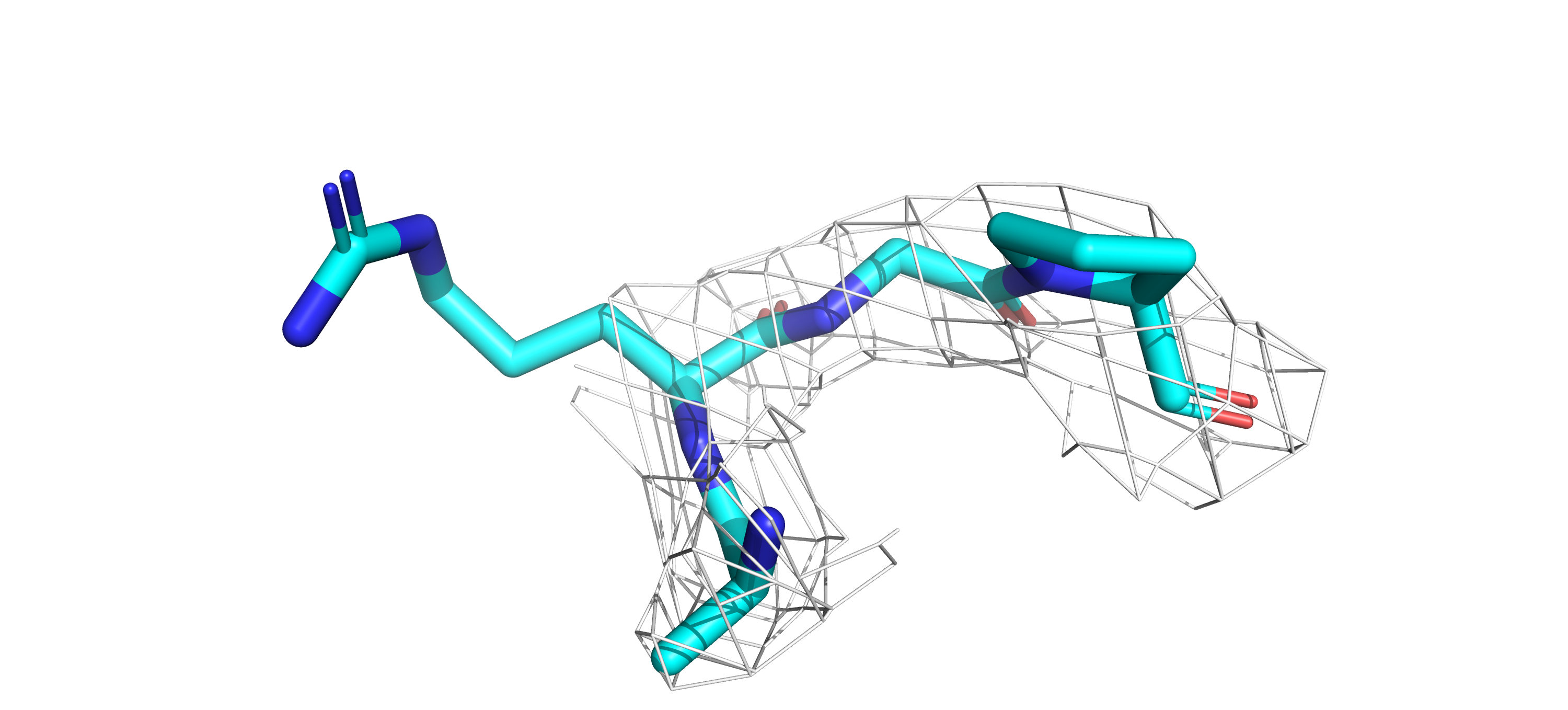
Посмотрела на связи который образует остаток глутамата в конформации А: рисунок 18. Сама аминокислота входит в состав
альфа-спирали, но там в образовании связей участвуют атомы вне радикала, положение которых одинаково
в обеих конформациях, поэтому смотрела только на связи радикала с другими группами белка. Есть водородные связи
с аргинином (длины 3.8 и 4.0, конечно, больше стандартных 3.5Å, но ничего ближе я не нашла).
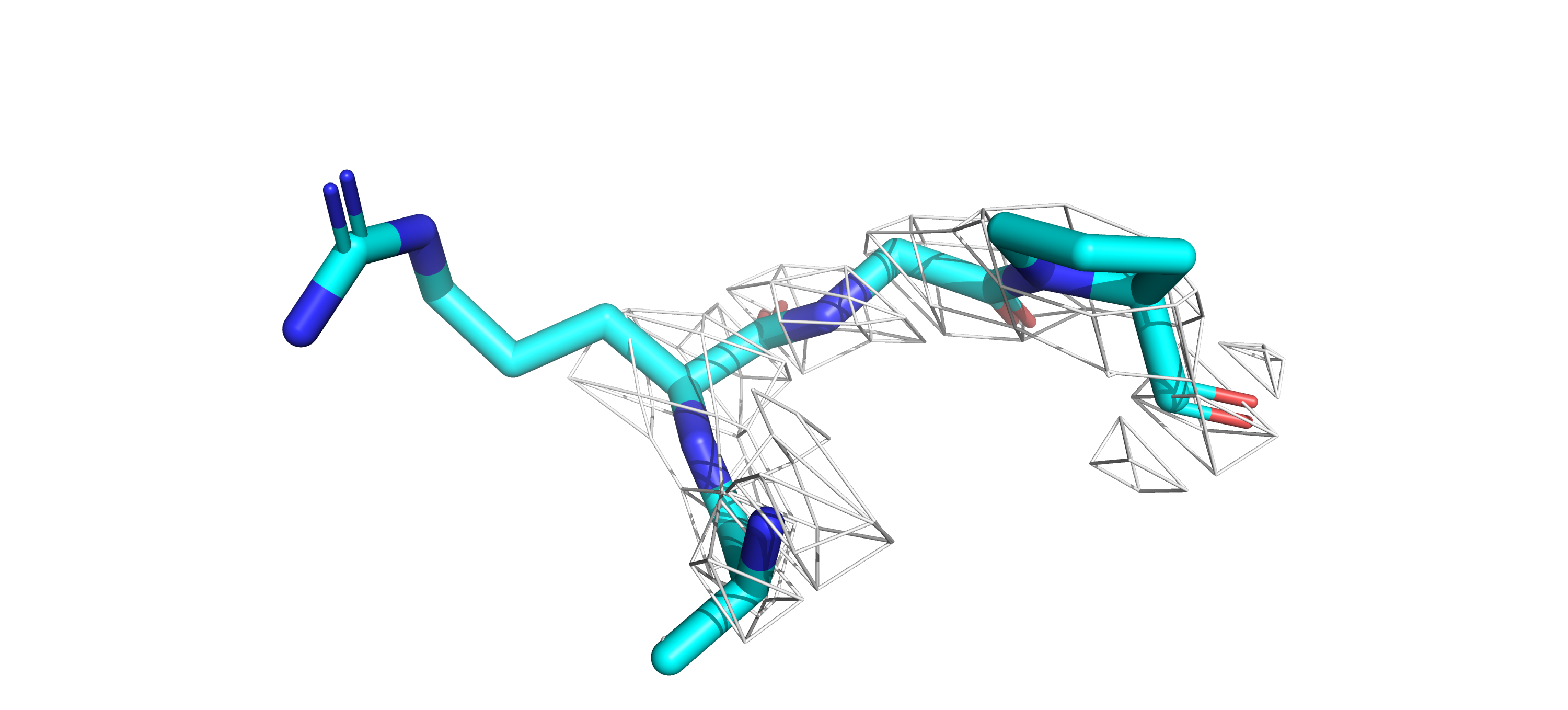
Далее изучила альтернативную конформацию: рисунок 19. Есть водородные связи с серином и треонином (для серина
длина в 4.5Å, конечно, тоже вызывает сомнения).
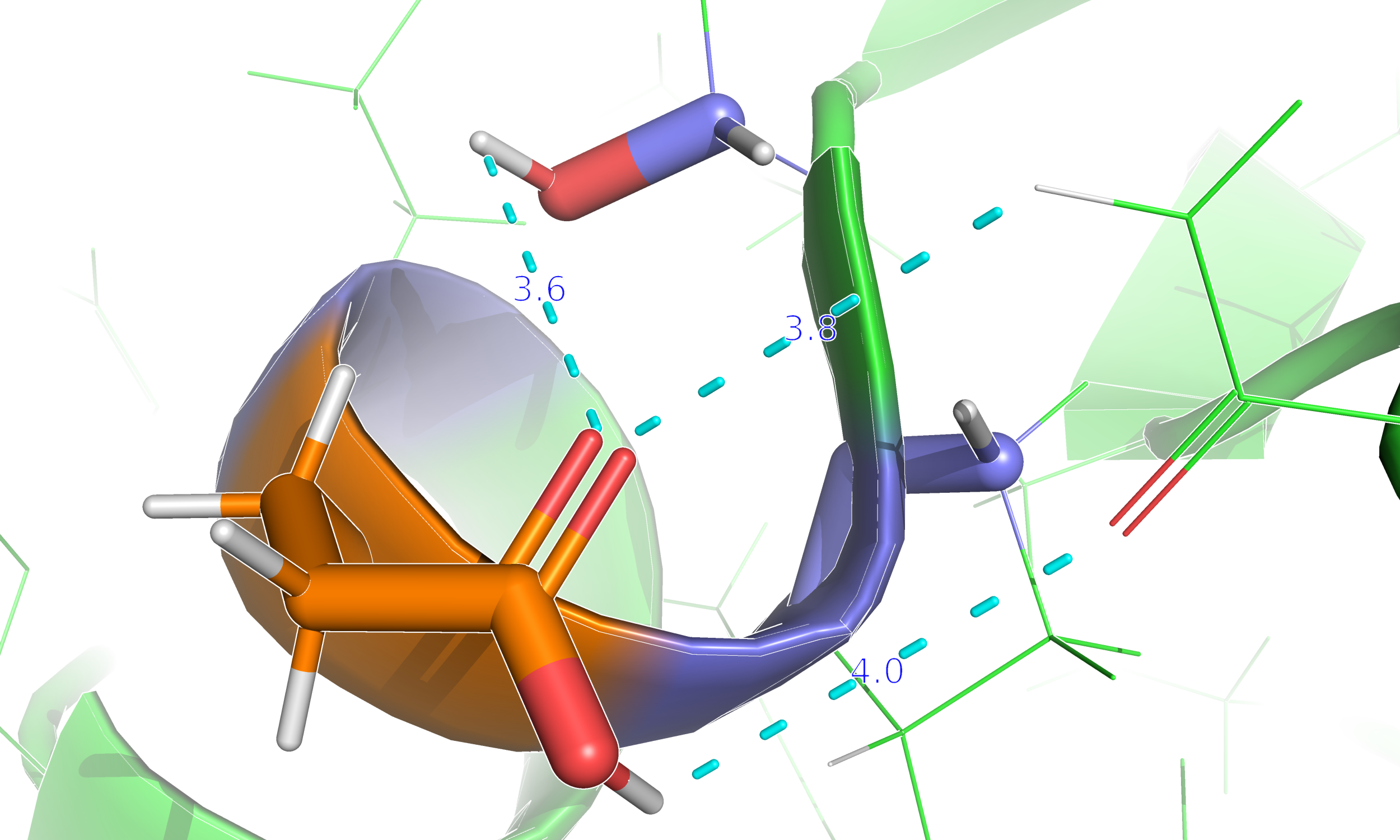

В целом, остатки одинаково стабилизированы в обеих конформациях. Хотя, возможно, распределение occupancy в 0.6 и 0.4 между A и B альтлоками, соответственно, было более логичным.
Задание 4. B-фактор
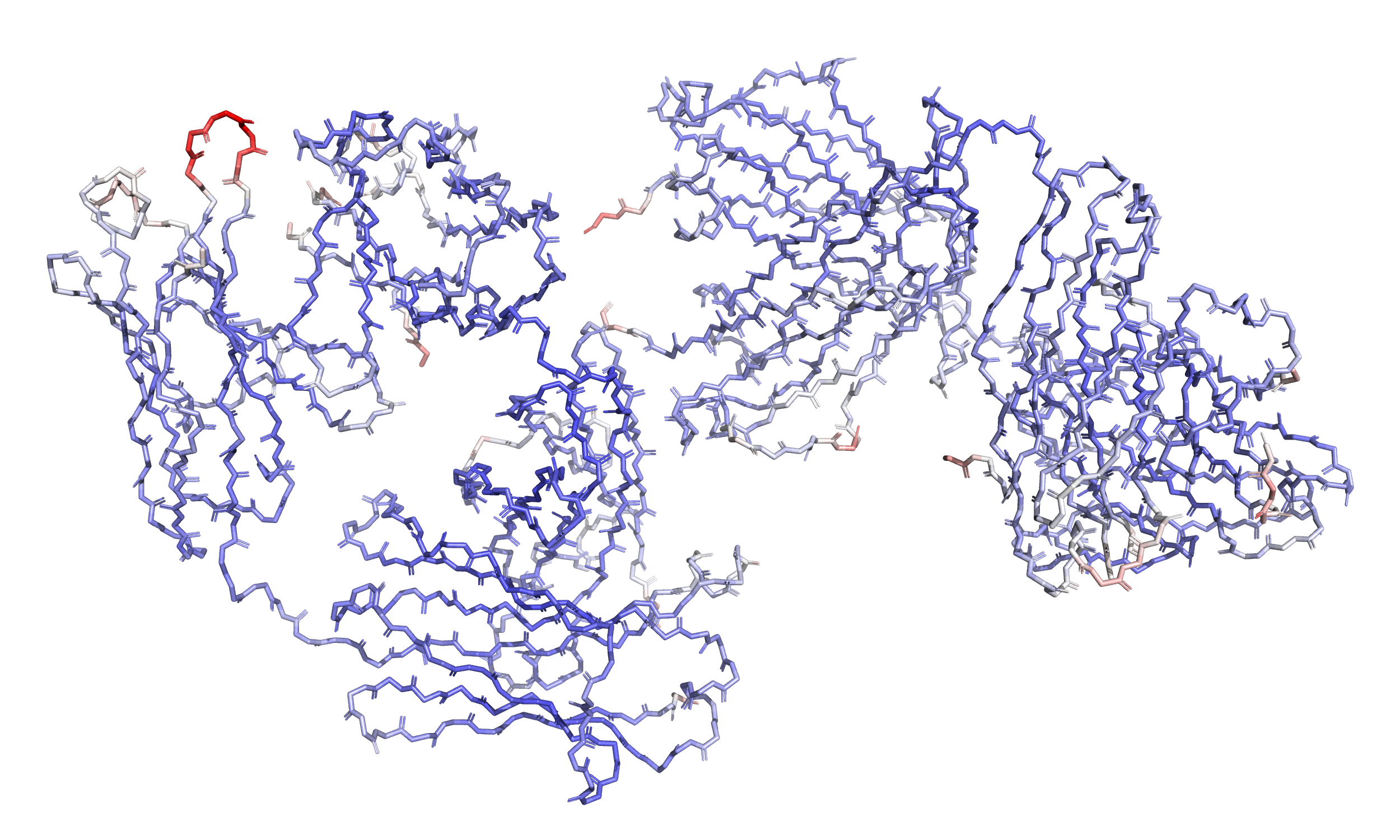
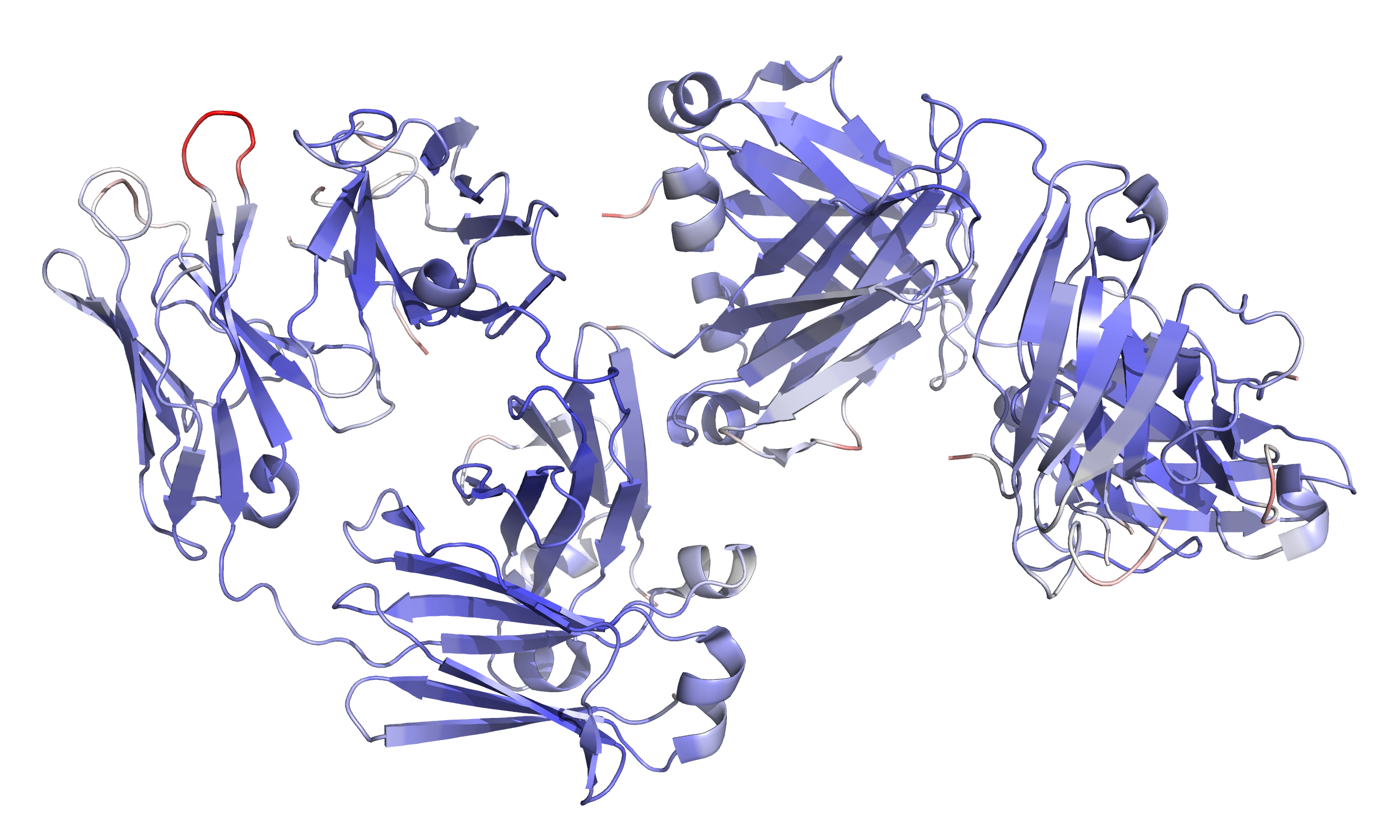
Продлжаю работать с моделью 8FO4. Покрасила атомы остова по B-фактору (рисунок 20):
spectrum b, blue_white_red, backbone
B-фактор отражает "термическую" подвижность атома, то есть неопределенность в положении атома, которая обычно связана с динамической гибкостью региона белка. В данном случае, у нас большая часть регионов синие - с низким B-фактором - то есть это очень стабильные участки структуры. На участках остова расположенных на поверхности есть несколько красных групп с высокой подвижностью (красные). Отображение cartoon (рисунок 21) показало, что синие регионы - элементы бета-листов - стабилизированных вторичных структур, неустойчивые же участки принадлежат не формирующим вторичных структур поверхностным аминокислотам.
Далее покрасила по B-фактору все атомы:
spectrum b, blue_white_red
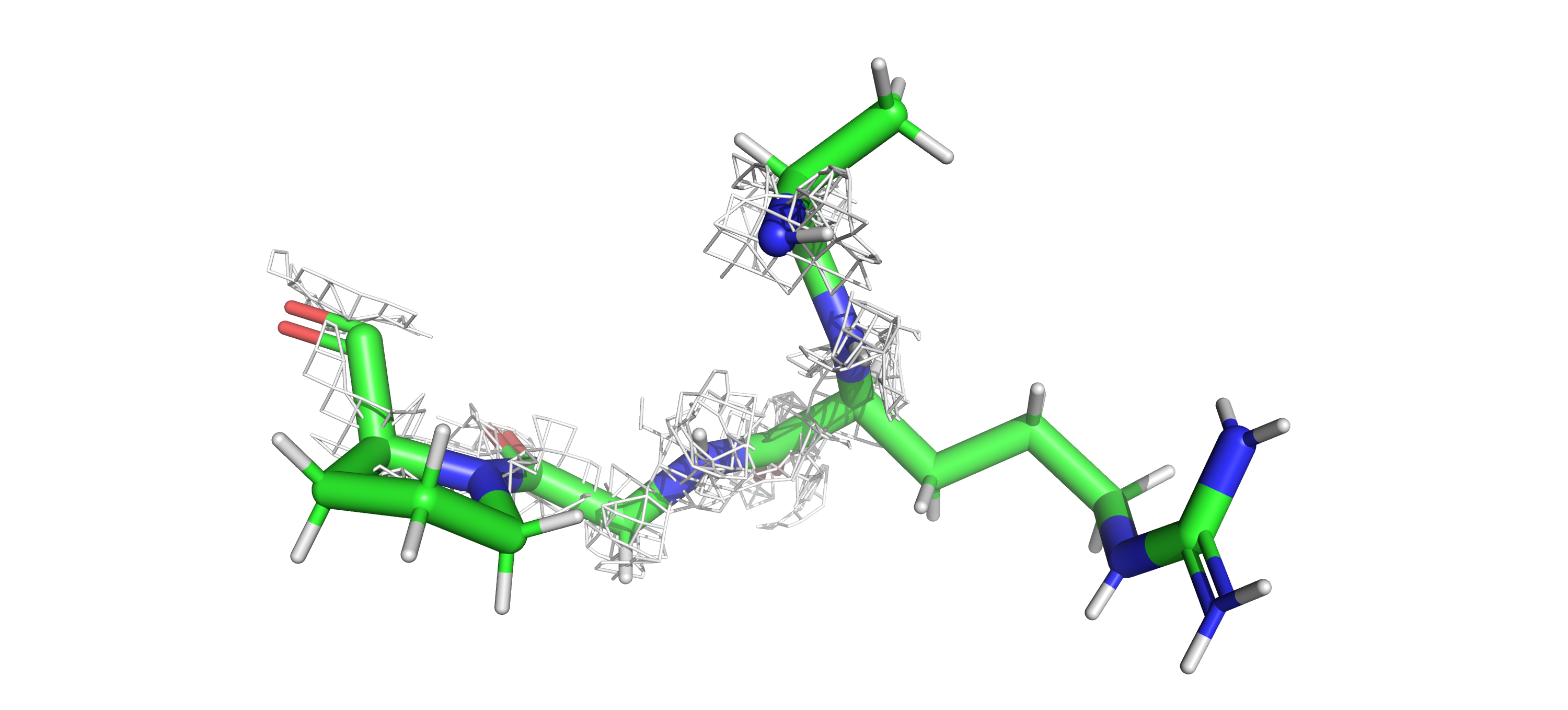
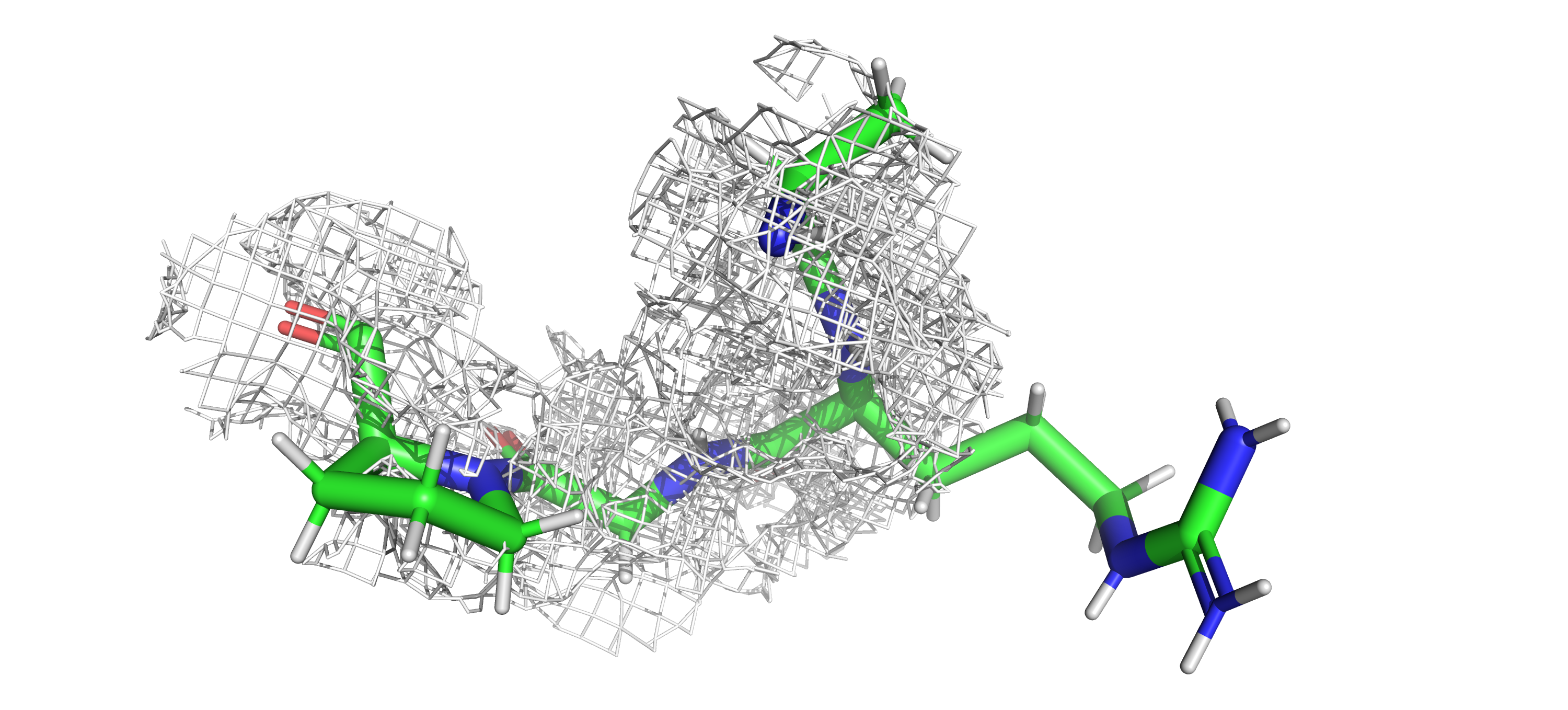
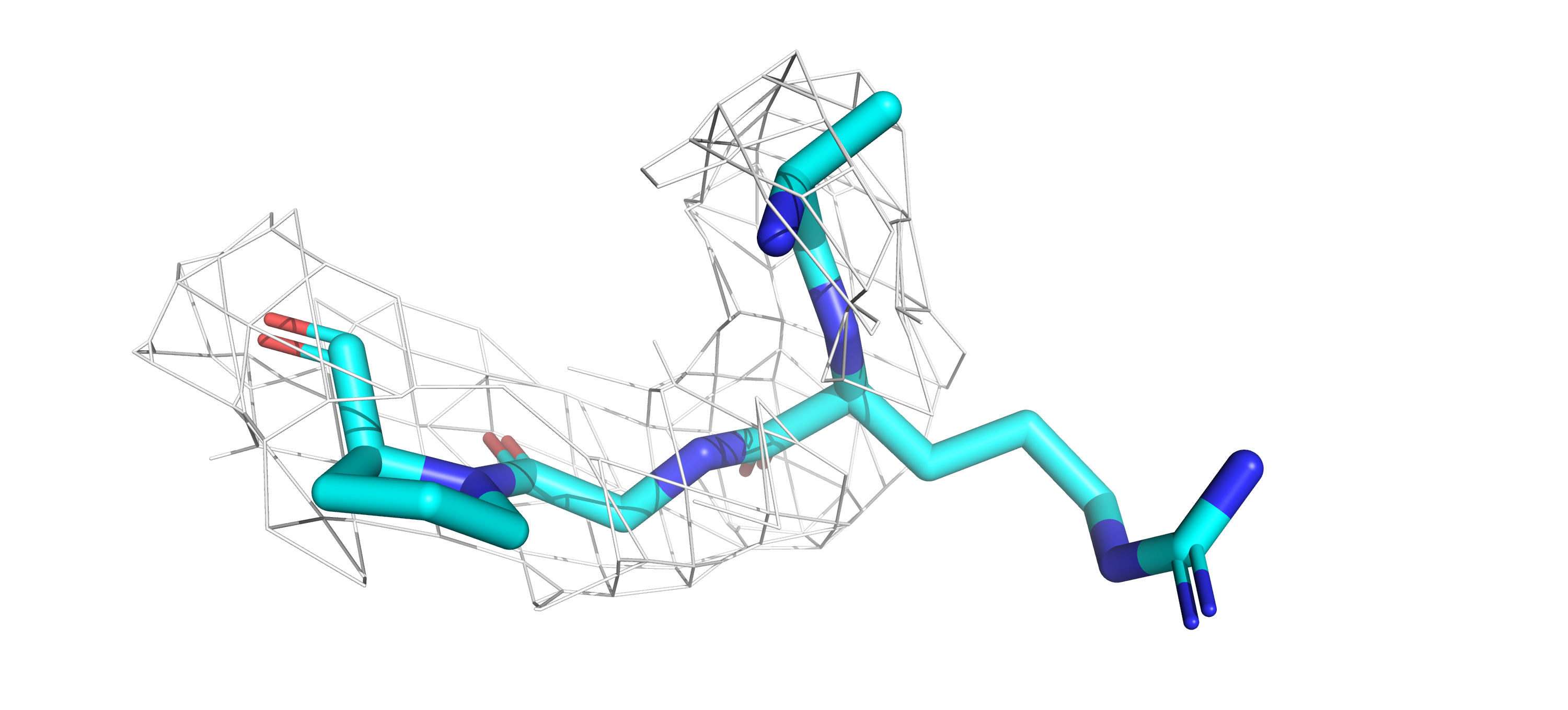
Изучала 44-ый остаток лизина на цепи C (серия визуализаций ЭП на разных уровнях показана на рисунках 22-24):
isomesh m0, 8FO4_2fofc, 0, (sele), carve=2 isomesh m1, 8FO4_2fofc, 1, (sele), carve=2 isomesh m2, 8FO4_2fofc, 2, (sele), carve=2
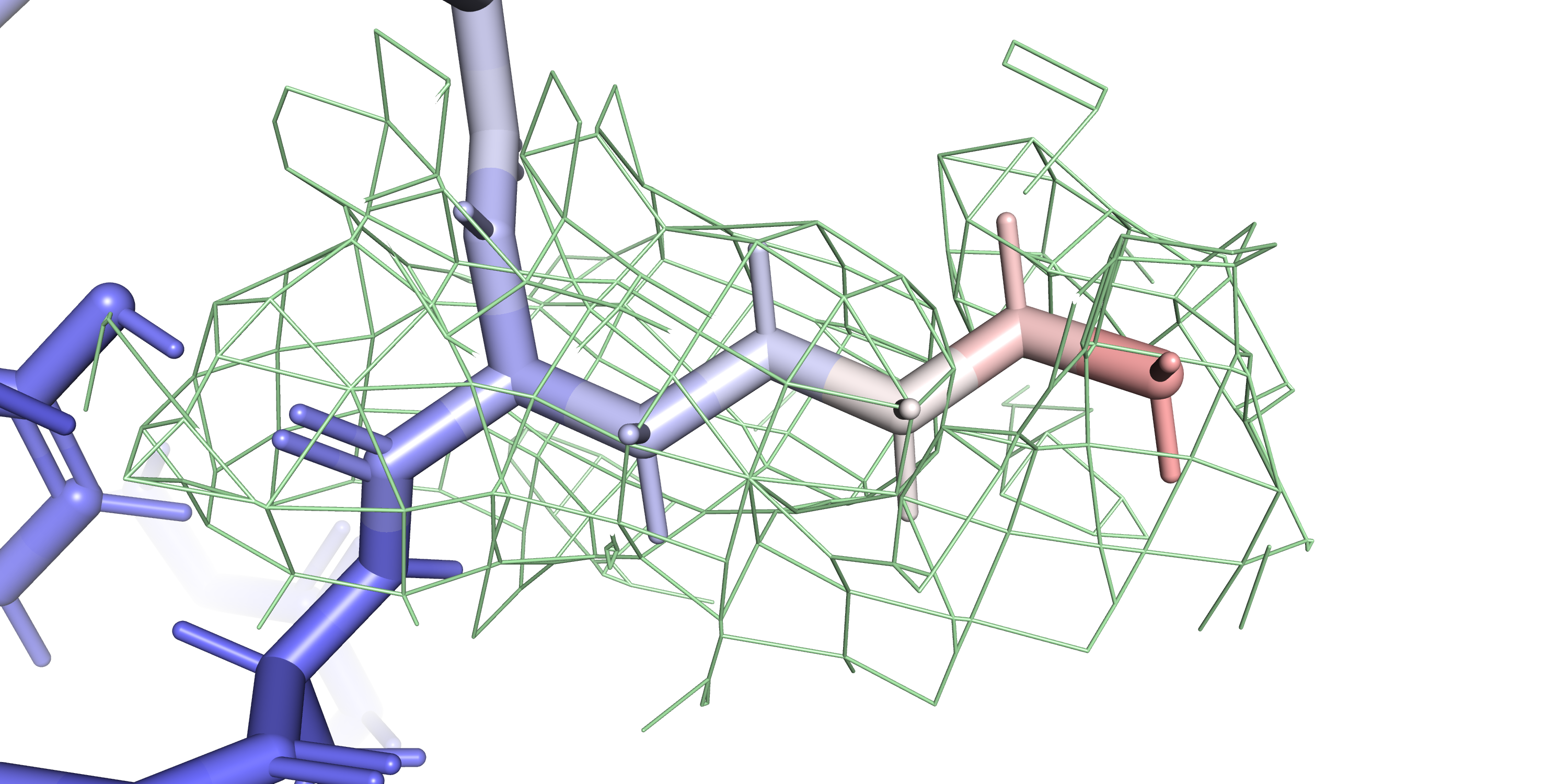

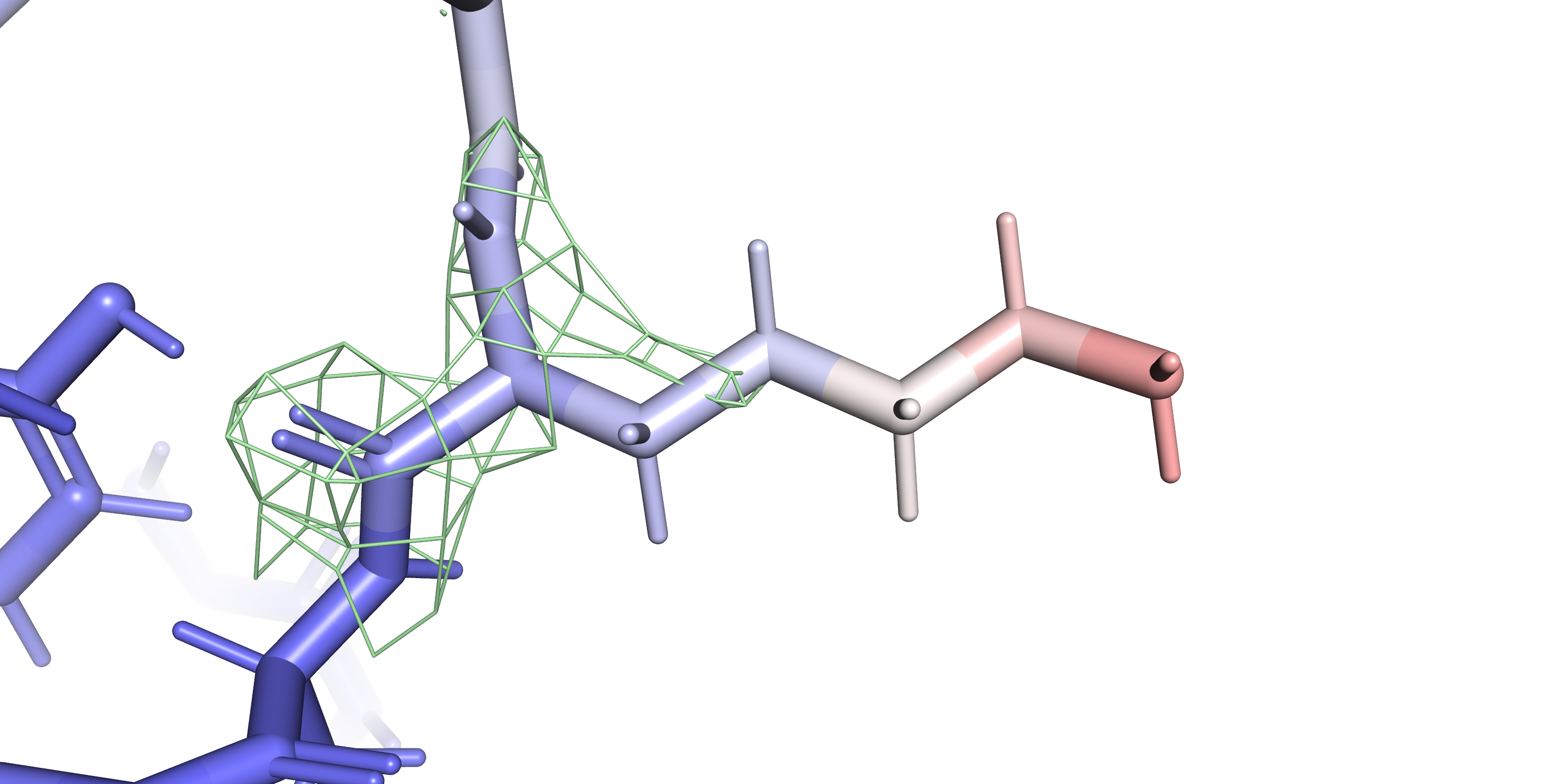
Видно, что наблюдения электронной плотности на разных уровнях подрезки зависят от B-фактора рассматриваемых атомов. Уже по B-фактору видно, что подвижность атомов радикала аминокислоты удаляется по мере удаления от остова (для амино-группы окраска даже становится красной). Для областей с высоким B-фактором ЭП размывается по мере повышения уровня подрезки, при этом на участках с низким B-фактором ЭП стабильна.