 Вконтакте
Вконтакте allakarpova@kodomo.fbb.msu.ru
allakarpova@kodomo.fbb.msu.ru vseokeyboss@gmail.com
vseokeyboss@gmail.comСравнение матриц аминокислотных замен.
Матрицы весов аминокислотных замен позволяют оценить насколько хорошо то или иное выравнивание.
Они строятся на основе баз данных аминокислотных последовательностей. Положительные числа в матрице указывают на
высокую вероятность такой замены, отрицательные - на низкую.
Существуют несколько матриц. Матрица BLOSUM62 создана по базе данных BLOCKS, где собраны в основном цитоплазматические белки.
Число 62, так называемый порог кластеризации, показывает, начиная с какого процента схожести, последовательности считаются идентичными, их объединяют в кластеры
и рассматривают в едином кластере, чтобы не сдвинуть вероятности.
Мембранные белки обладают иным содержанием аминокислот, следовательно матрица BLOSUM62 плохо описывает выравнивания таких белков.
Так была создана матрица PHAT, основанная на встречаемости аминокислот в трансмембранных белках.
На основе базы данных BLOCKS мною была реконструированна матрица BLOSUM. Все рассчеты и последовательно полученные матрицы
можно посмотреть здесь. В целом матрицы (моя и BLOSUM62) очень схожи. Положительные значения одной соответствуют положительным значениям другой,
отрицательные - отрицательным. Для наглядности рассмотрим веса замен аспарагина на другие аминокислоты по трем матрицам (Табл.1)
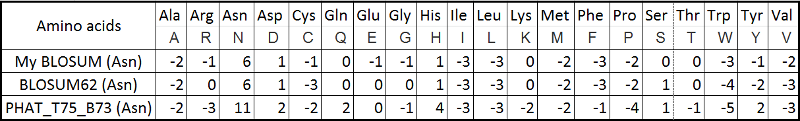 Табл.1. Сравнение весов замен аминокислот из матриц BLOSUM62, PHAT и из личной.
Табл.1. Сравнение весов замен аминокислот из матриц BLOSUM62, PHAT и из личной.
- Вес замены аспарагина на самого себя по таблице PHAT отличается от веса по таблицам BLOSUM (62 и моей) почти в 2 раза (11 и 6 соответственно). Таблица PHAT составляется для трансмембранных белков, где, видимо, сильно сдвинута частота встречаемости и замены аминокислот и аспарагина в том числе. Видимо,наблюдаемая частота встречаемости аспарагина больше ожидаемой, значит аспарагин чаще заменяется на самого себя, вот и вес замены больше. Вообще трансмембранные белки консервативны в своей структуре, поэтому важно не принебрегать такой полярной и незаряженной аминокислотой, как аспарагин.
- Замена на схожие аминокислоты. Аспарагин по радикалу относится к незаряженным полярным аминокислотам. Вес замены на крайне похожий глютамин по матрицам blosum равен 0, а по матрице PHAT на 2 единицы больше. Опять-таки это можно связать с общей консервативностью структуры мембранных белков, для которых важно, чтобы полярная и незаряженная аминокислота заменилась на похожую да и ещё с такой же функциональной группой. Странно, но замена на Тирозин оказывается выгодной по матрице PHAT (2) и не выгодной по моей матрице (-1) и матрице blosum62 (-2). Asn и Tyr объединяет полярный радикал, но разнит ароматическое строение тирозина. Видимо, в трансмембранных белках важна полярность/гидрофобность, а не алифатичность/ароматичность. Замена на серин одинакова по blosum62 и PHAT (1) и равна 0 по моей матрице. На треонин аспарагин хуже заменять в трансмембранных белках, чем в цитоплазматических, может быть полярную -ОН группу треонина закрывает -СН3, что делает его чуть больше гидрофобным, чем ASN. Можно заметить, что на гистидин хорошо заменяется аспарагин. Я думаю, это связано с тем, что HIS незаряжен при клеточном рН (рК=6), следовательно, является полярной и незаряженной аминокислотой.
- Замена на противоположные по химическим свойствам аминокислоты. Замена на PHE (гидрофобная ароматика) не выгодна как по матрицам BLOSUM (-2), так и по PHAT (-4). По PHAT наименее выгодна, т.к. гидрофобная аминокислота не должна быть в гидрофильной части трансмембранного белка (где обычно располагается полярный аспарагин). Вредны для трансмембранных замены и на +заряженные лизин и аргинин. Вес замены по матрцам BLOSUM равен 0 (по моей матрице вес замены на аргинин -1), а по PHAT вес замены на лизин и аргинин равен -2 и -3 соответственно. Появление + вместо нулевого заряда может помешать (почти всегда)белку встроиться в мембрану и выполнять свою функцию. Для цитоплазматических это может быть не так критично, если замена произошла не в стратегически важном месте, конечно.
Вообще различия между моей матрицей и матрицей BLOSUM62 незначительны, если судить по заменам аспарагина. Для 12 аминокислот веса замен одинаковы, для всех остальных отличаются на единицу, за исключением цистеина. BLOSUM62 составлась давно, много белков с тех пор описано и геномов секвенировано, из-за новых данных изменилась встречаемость аминокислот. Расхождения возможны ещё и потому, что мы не рассчитывали вес для ГЭПов и B,Z,X, из-за чего вероятности исказились.
Сравнение выравниваний, полученных для коротких мутантов вручную и построенных классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана
Алгоритм Нидлмана-Вунша направлен на выравнивание целой последовательности с нахождением наибольшего процента схожести. По этому алгоритму работает команда needle пакета EMBOSS.
Алгоритм Смита-Ватермана выравнивает куски последовательностей. Этот алгоритм реализует команда water пакета EMBOSS.
Обе команды имеют параметры штрафа за один ГЭП (от 0 до 100) и за последующие ГЭПы, если они идут непрерывно (от 0 до 10).
По умолчанию первый параметр имеет значение 10, второй - 0,5.
Для подсчета веса выравнивания по умолчанию используется матрица BLOSUM62.
Мною были получены при помощи скрипта evolve_protein.pl 3 изменных последовательности из 20 аминокислот из последоватльности белка ybbd_bacsu.
- Change=0.6 Replace=0.6
Выравнивание вручную:
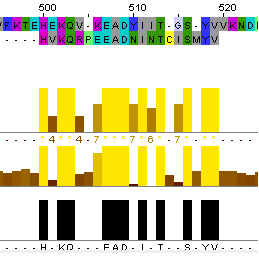
1) Length - 20 (645 в целом) 2) Identity - 55% (1,7% в пересчете на длину белка) 3) Similarity - 55% (1,7% в пересчете на длину белка) 4) Score - 32.
Выравнивание needle
YBBD_BACSU 451 KGSRILIVAPYEEQTASIEQTIHDLIKRKKIKPVSLSKMNFASQVFKTEH 500 | generations=1 1 -------------------------------------------------H 1 YBBD_BACSU 501 EKQ-VKEADYIIT--GSYVVKNDPVVNDGVIDDTISDSSKWATVFPRAVM 547 .|| .:|||.|.| ..|| generations=1 2 VKQRPEEADNINTCISMYV------------------------------- 20 1)Length: 645 2)Identity: 10/645 ( 1.6%) 3)Similarity: 11/645 ( 1.7%) 4)Gaps: 628/645 (97.4%) 5)Score: 23.5Выравнивание water
YBBD_BACSU 500 HEKQ-VKEADYIIT 512 |.|| .:|||.|.| generations=1 1 HVKQRPEEADNINT 14 1)Length: 14 2)Identity: 8/14 (57.1%) 3)Similarity: 9/14 (64.3%) 4)Gaps: 1/14 ( 7.1%) 5)Score: 24.0 - Change=0.6 Replace=0.8
Выравнивание вручную:

1) Length - 21 (642 в целом) 2) Identity - 43% (1,4% в пересчете на длину белка) 3) Similarity - 43% (1,4% в пересчете на длину белка) 4) Score - 24.
Выравнивание needle
YBBD_BACSU 351 SVTSLKEEQKFARVIQALKEAVKNGDIPEQQINNSVERIISLKIKRGMYP 400 |:|.|...::..|.|.::.. generations=1 1 -----KKEGKIQFMLMQLHEGIETM------------------------- 20 1)Length: 642 2)Identity: 5/642 ( 0.8%) 3)Similarity: 10/642 ( 1.6%) 4)Gaps: 622/642 (96.9%) 5)Score: 21.0Выравнивание water
YBBD_BACSU 356 KEEQKFARVIQALKEAVK 373 |:|.|...::..|.|.:: generations=1 1 KKEGKIQFMLMQLHEGIE 18 1)Length: 18 2)Identity: 5/18 (27.8%) 3)Similarity: 10/18 (55.6%) 4)Gaps: 0/18 ( 0.0%) 5)Score: 24.0 - Change=0.4 Replace=0.8
Выравнивание вручную:

1) Length - 20 (644 в целом) 2) Identity - 70% (2,2% в пересчете на длину белка) 3) Similarity - 75% (2,3% в пересчете на длину белка) 4) Score - 65.
Выравнивание needle
YBBD_BACSU 101 QTVQLTDDYQKASPKIPLMLSIDQEGGIV-TRLGEG-TNFPGNMALGAAR 148 |.||||||| ||:.|| .|| generations=1 1 --------------------SCDQEGGIVRTRMIEGFLNF---------- 20 1)Length: 644 2)Identity: 14/644 ( 2.2%) 3)Similarity: 15/644 ( 2.3%) 4)Gaps: 626/644 (97.2%) 5)Score: 49.0Выравнивание water
YBBD_BACSU 121 SIDQEGGIV-TRLGEG-TNF 138 |.||||||| ||:.|| .|| generations=1 1 SCDQEGGIVRTRMIEGFLNF 20 1)Length: 20 2)Identity: 14/20 (70.0%) 3)Similarity: 15/20 (75.0%) 4)Gaps: 2/20 (10.0%) 5)Score: 49.0
