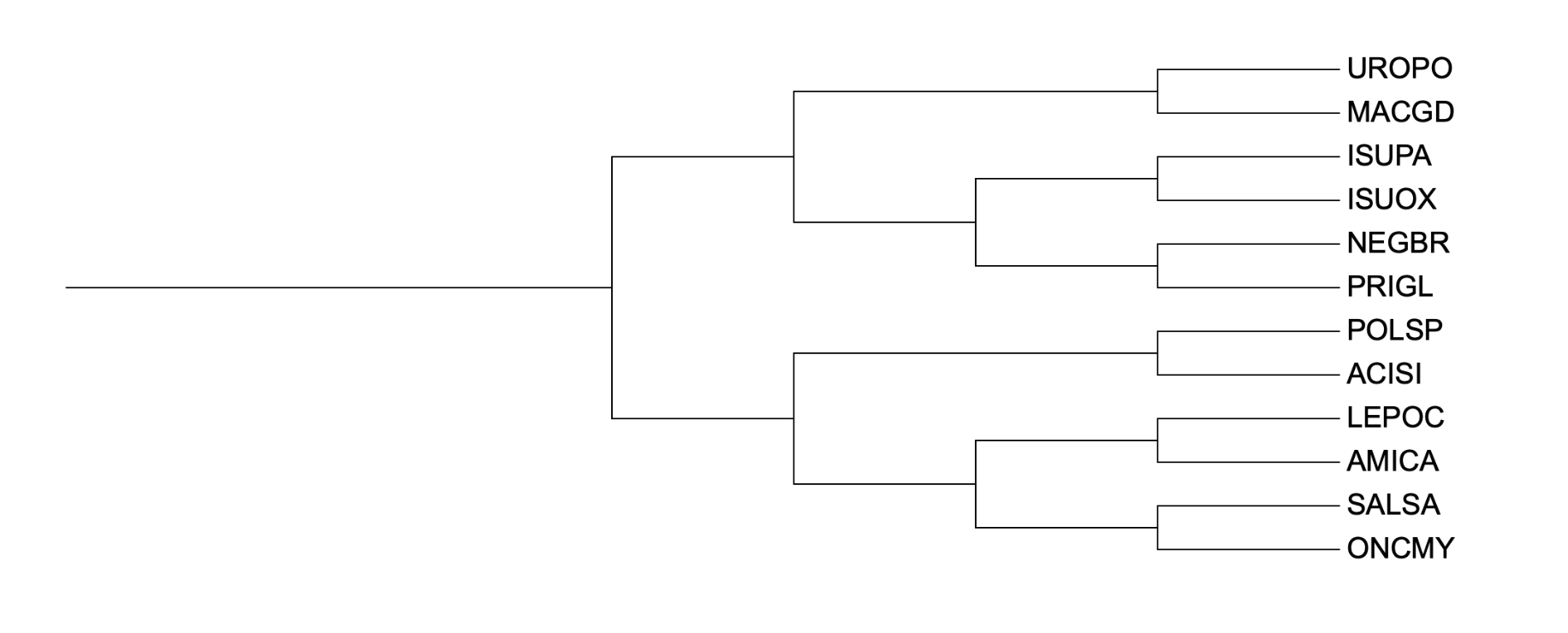
Целью данного практикума была реконструкция филогенетических деревьев для выбранных видов животных на основе последовательностей цитохрома B. Для этого были использованы методы выравнивания последовательностей, преобразования форматов и построения деревьев с помощью программ FastME и IQ-TREE. Полученные деревья сравнивались с исходной таксономической классификацией для оценки точности реконструкции.
На первом этапе последовательности цитохрома B были загружены из базы данных Swiss-Prot с использованием программы EMBOSS (команда `seqret`). Затем последовательности были выровнены с помощью программы Muscle. Для реконструкции деревьев программа FastME требует формат выравнивания phylip-relaxed, поэтому выравнивание было преобразовано в этот формат с использованием библиотеки BioPython.
Филогенетические деревья были построены двумя методами:
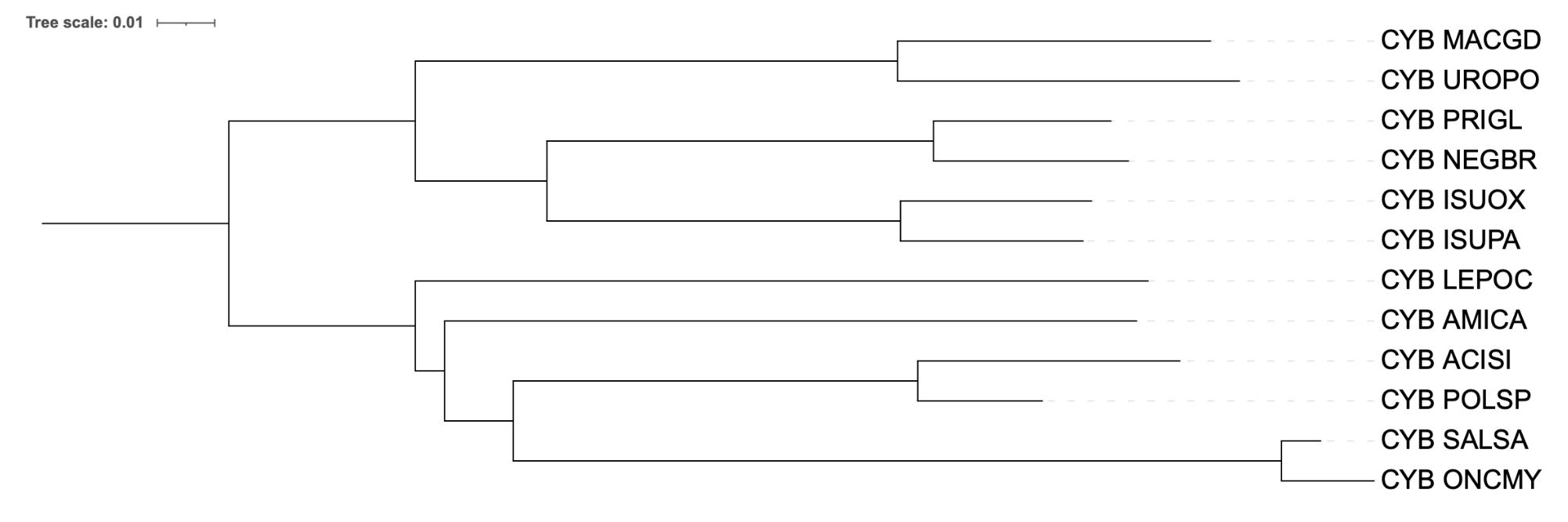
На рисунках ниже представлены реконструированные деревья:
В таксономическом дереве есть клада состава клада {LEPOC, AMICA}, которая не выделяется ни в одной из реконструкций, что указывает на расхождение между филогенетической реконструкцией и принятым таксономическим положением этих двух видов. Вместо нее на реконструированных деревьях в этой кладе LEPOC должен был иметь родственный вид, являющийся предком AMICA
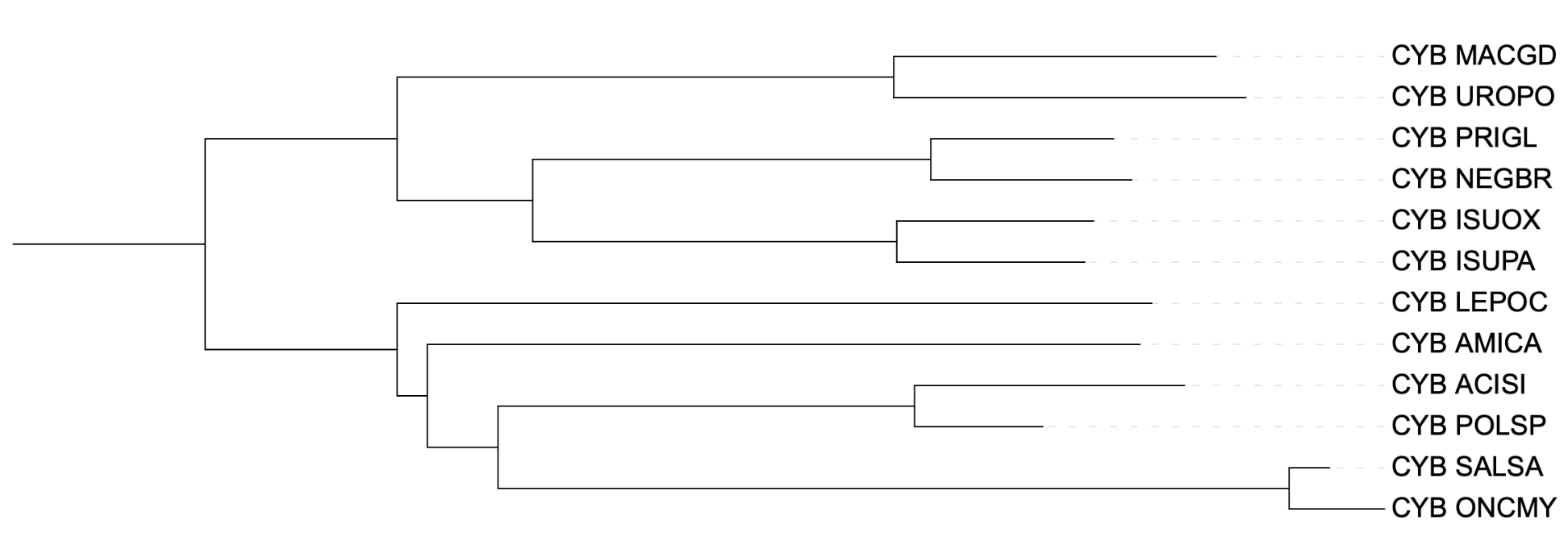
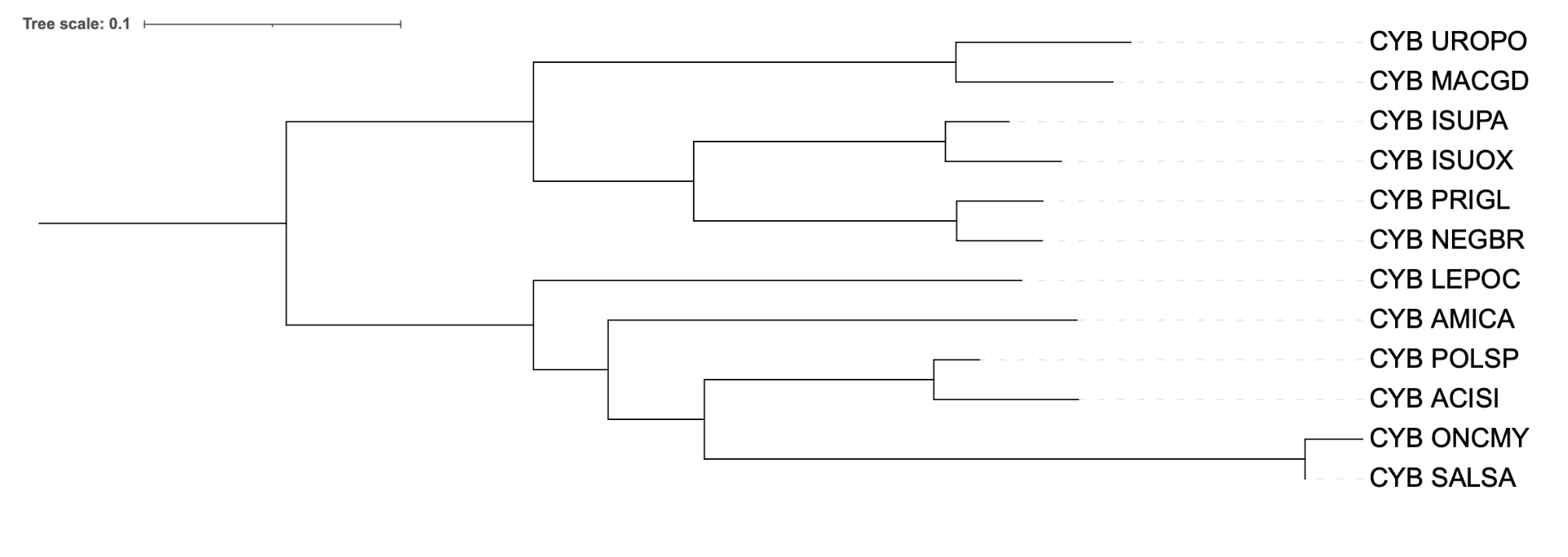
Анализ показал, что деревья, построенные с использованием модели p-distance (FastME), наиболее точно отражают таксономию, однако были замечены некоторые расхождения, особенно в позиционировании LEPOC и AMICA. Дерево IQ-TREE также демонстрирует отклонения от таксономии, группы расположены совсем не так, в модели MtRev мы видим примерно те же ошибки, что в случае p-distance.