Моделирование и реконструкция эволюции гена
C помощью графического редактора Paint было нарисовано дерево
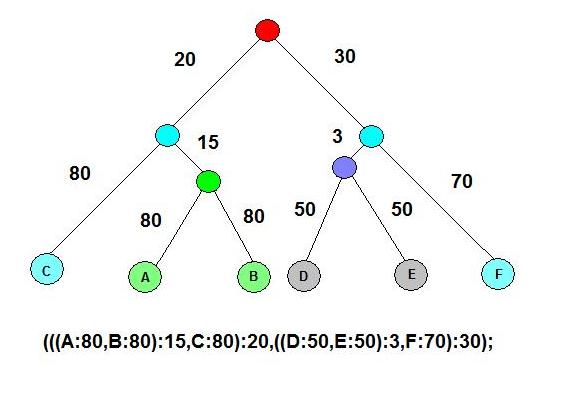 Данное дерево является бинарным, не ультраметрическим (так как растояния от корня
до листьев не одинаковые). Корнем является ген yahk.
Данное дерево является бинарным, не ультраметрическим (так как растояния от корня
до листьев не одинаковые). Корнем является ген yahk.
Описание ветвей дерева как разбиения множества листьев
A B C D E F
. . * * * *
. . . * * *
. . . * * .
Получение искусcтвенных мутантных последовательностей с помощью
программы msbar пакета EMBOSS
Длина гена 1050 нуклеотидов
Формула для пересчета расстояний в число мутаций:
N - число мутаций
S - длина гена
а - количество замен на 100 н.п
Коэффициент 1.33:
При генерации мутантных последовательностей программа msbar осуществляет
с равной вероятностью замены нуклеотидов на один из четырех возможных вариантов.
Например, нуклеотид A может быть заменен не только на G, C или T, но и на A с
той же вероятностью. Поэтому, значение параметра count (общее количество
вносимых замен) в 4/3 раза превышает количество замен, приводящих к изменению
нуклеотидного состава последовательности. Следовательно, для введения в
последовательность n замен необходимо задать значение параметра count
равным 4/3 n = 1.33n.
Мутантные последовательности были полученны с помощью скрипта:
msbar 1uufn.fasta abc.fasta -point 4 -count 279 -auto
msbar abc.fasta c.fasta -point 4 -count 1117 -auto
msbar abc.fasta ab.fasta -point 4 -count 210 -auto
msbar ab.fasta a.fasta -point 4 -count 1117 -auto
msbar ab.fasta b.fasta -point 4 -count 1117 -auto
msbar 1uufn.fasta def.fasta -point 4 -count 419 -auto
msbar def.fasta de.fasta -point 4 -count 43 -auto
msbar de.fasta d.fasta -point 4 -count 698 -auto
msbar de.fasta e.fasta -point 4 -count 698 -auto
msbar def.fasta f.fasta -point 4 -count 978 -auto
Реконструкция дерева
Файл с выравниванием листьев дерева : listya.fasta
Реконструкция дерева алгоритмом максимального правдоподобия программы fdnaml
fdnaml listya.fasta -ttratio 1 -auto
результат: fdnaml_results.txt
+----------------------b
|
| +----------------------f
| +--4
| +------------------3 +---------------e
| | |
1-------2 +-------------d
| |
| +----------------------c
|
+-------------------------a
(b:0.76281,(((f:0.76567,e:0.51260):0.09166,d:0.45527):0.62289,c:0.76458):0.25596,a:0.87273);
Реконструкция дерева алгоритмом Neighbor-joining:
fdnadist lystya.fasta -ttratio 1 -auto
fneighbor lystya.fdnadist -auto
результат: neighbor_joining_results.txt
+-------------b
!
! +-------------c
1------2
! ! +------d
! +--------------3
! ! +---------e
! +---4
! +---------------f
!
+------------------a
(b:0.71780,(c:0.71649,(d:0.36992,(e:0.50426,f:0.79928):0.19289):0.72491):0.35823,a:0.92998);
Реконструкция дерева алгоритмом UPGMA:
fdnadist lystya.fasta -ttratio 1 -auto
fneighbor lystya.fdnadist -outfile list.fneighbor
результат:upgma_results.txt
+------------------------a
+---3
+-------4 +------------------------b
! !
! +----------------------------c
--5
! +---------------d
! +---1
+----------------2 +---------------e
!
+-------------------f
(((a:0.82389,b:0.82389):0.12542,c:0.94931):0.26981,((d:0.53497,e:0.53497):0.13073,f:0.66570):0.55342);
Сравнение деревьев:
| ABCDEF |
real |
fdnaml |
Neighbor-joining |
UPGMA |
| . . . . * * |
- |
+ |
+ |
- |
| . . . * * * |
+ |
+ |
+ |
+ |
| . . * * * * |
+ |
+ |
+ |
+ |
| . . . * * . |
+ |
- |
- |
+ |
Как видно из таблицы, больше всего на исходное дерево похоже дерево, реконструированное по методу UPGMA.
Деревья, построенные по алгоритму fdnaml и Neighbor-joining, вместо ветви, содержащей листья D и E,
имеют ветвь с листьями E и F. Возможно, это связано с тем, что между ветками F и (DE)
всего 3 замены на 100 пар нуклеотидов.
Алгоритм UPGMA реконструирует ход эволюции основываясь на предположении о справедливости гипотезы "молекулярных
часов" (скорость накопления мутаций поcтоянна). Вследствие этого суммарные расстояния от корня дерева
до каждого из его листьев равны друг другу. Возможно, более высокое сходство дерева,
реконструированного по методу UPGMA, с исходным деревом связано с тем, что, помимо матриц расстояний,
данный алгоритм использует дополнительное предположение о справедливости гипотезы "молекулярных часов".
Также стоит отметить, что алгоритм UPGMA, в отличии от NJ и ML строит укорененное дерево
Методы NJ(эвристический) и ML(переборный) учитывают различия в скорости накопления мутаций в разных последовательностях.
Это дает определенные преимущества при анализе выборок реальных последовательностей. Но для рассмотренной
модели эволюции имеют место значительные расхождения между истинными эволюционными расстояниями и их оценками.
Например, истинные расстояния от точки расхождения последовательностей А и B равны 80, метод NJ оценил их равными
93 и 80 соответственно, а метод ML - 87 и 76 соответственно. Алгоритм ML, в отличие от NJ, позволил оценить
доверительные интервалы длин ветвей уровня значимости 0.01 .
Следует отметить, что метод NJ справился с реконструкцией дерева значительно быстрее, чем метод ML.
Для выборки из шести последовательностей различия во времени работы составляют секунды, однако для выборок
реально существующих последовательностей большего объема различия по времени работы могут оказать влияние на выбор алгоритма.
на страницу IV семестра
на главную