Практикум 8. Нуклеотидный BLAST
Задание 1.
В практикуме 7 я выбара организм Pusa sibirica, но в ncbi не окзалалсь genome data view, поэтому для данного задания выбрала другой организм - Canis lupus familiaris(пудель)
Выбранный CDS
1.Идентификатор нуклеотидной записи, откуда был взят фрагмент: NC_049258.1
2.Координаты фрагмента: 15,145,275 - 15,164,212
3.Длинa CDS: 15,778 nt
4.В этот участок попал ген: FASTKD2.Ген FASTKD2 кодирует белок, который играет важную роль в регуляции митохондриальной функции, метаболизме и апоптозе.
5.На картинке изображено несколько вариантов первичных транскриптов, которые могут получаться с исходного гена. В данным случае этих вариантов восемь.
ссылка на файл с последовательностью в формате fasta
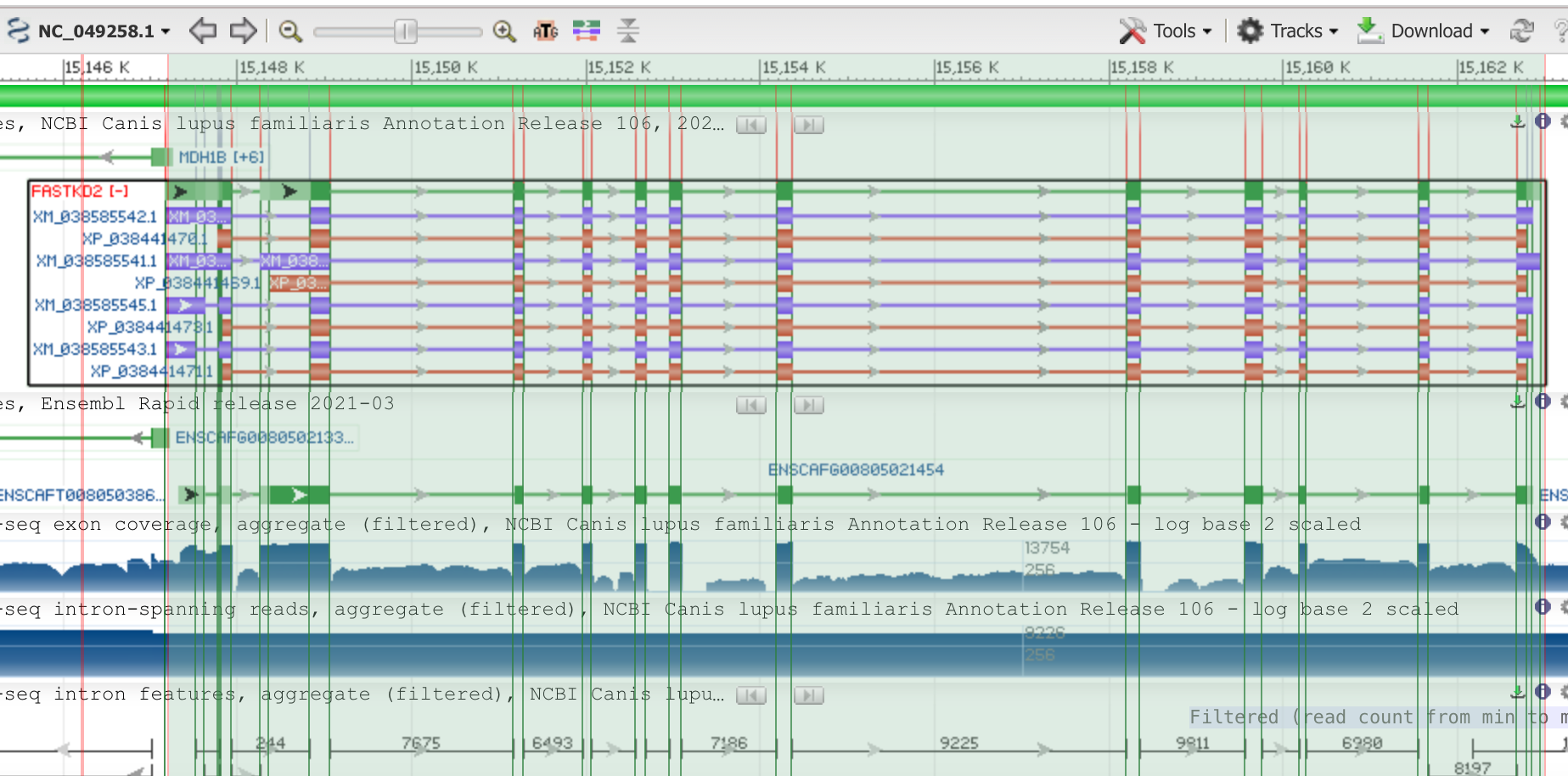
Рис.1 Данный ген FASTKD2 закодирован между 15,147,177..15,162,954, имеющий длину 15,778 nt. Красным обозначены CDS; Фиолетовым - mRNA; Зеленым - сам ген, , широкие области соответствуют экзонам, а узкие - интронам. Далее идут данные по покрытию RNA-seq. Нижний блок содержит информацию о характеристиках интронов, полученную с помощью RNA-seq.
Поиск BLAST
Более дальним родственным таксоном Canidae( к которому относится Canis lupus familiaris) является семейство в отряда кошачьи (Felidae), поэтому решила выбрать его для сравнения
blastn: Этот алгоритм предназначен для сравнения нуклеотидных последовательностей. Он полезен для нахождения сходства между двумя нуклеотидными последовательностями, такими как ДНК или РНК: сравнивать геномы неблизкородственных организмов, а также устанавливать, к какому организму принадлежит тот или иной фрагмент генома.
Наше исследование: Длина слова - 11; Кол-во находок - 100; Находки - query cover:5-15% но больше чем у половины:0%, но зато процент идентичности варьируется 69-84%
megablast: Этот более быстрый и менее чувствительный вариант blastn и рекомендуется для сравнения больших нуклеотидных последовательностей. Его можно использовать, когда мы уверены, что наша последовательность найдется в каким-то конкретном организме. Это актуально, когда поиск проводится внутри одного и того же рода.
Наше исследование: Длина слова - 28; Кол-во находок - 26; Находки - query cover: 3-9%,у последних 4:0%, процент идентичности варьируется 81-91%
blastx: Этот алгоритм используется для аннотации последовательности, так как последовательность переводится из нуклеотидной в аминокислотную и поиск проводится по белковой базе данных.
Наше исследование: Длина слова - 5; Кол-во находок - 17; Находки - query cover: 13-14%,процент идентичности варьируется 59-68 %
tblastx: Данный алгоритм предназначен для сравнения в шести рамках - он транслирует обе нуклеотидные последовательности и ищет сходства во всех шести рамках. Его применяют для поиска таких гомологичных последовательностей, которые не были аннотированы как гены белков
Наше исследование: Вылезает ошибка, связанная со слишком большим использованием CPU. Я пыталась запускать эту программу более щадящими CPU параметрами, но все равно вылезала ошибка
Задание 2.
1.Индексация генома
makeblastdb -in GCF_011100685.1_UU_Cfam_GSD_1.0_genomic.fna -dbtype nucl
Вывод 8 файлов:
GCF_011100685.1_UU_Cfam_GSD_1.0_genomic.fna.ndb
GCF_011100685.1_UU_Cfam_GSD_1.0_genomic.fna.nhr
GCF_011100685.1_UU_Cfam_GSD_1.0_genomic.fna.nin
GCF_011100685.1_UU_Cfam_GSD_1.0_genomic.fna.njs
GCF_011100685.1_UU_Cfam_GSD_1.0_genomic.fna.not
GCF_011100685.1_UU_Cfam_GSD_1.0_genomic.fna.nsq
GCF_011100685.1_UU_Cfam_GSD_1.0_genomic.fna.ntf
GCF_011100685.1_UU_Cfam_GSD_1.0_genomic.fna.nto
Берем последовательности рРНК Escherichia coli для проведения локального поиска BLAST отдельно для каждой рРНК из файла
16S рРНК - находится в малой субъединице рибосомы у прокариот и играет важную роль в трансляции генетической информации. Она участвует в распознавании последовательности Шайн-Дальгарно на мРНК и стабилизации кодон-антикодонового взаимодействия на аминокислотном сайте рибосомы, а также также способствует формированию структуры рибосомы и связывается с белками малой субъединицы, что обеспечивает стабильность и функциональную активность рибосомы.
23S рРНК-находится в большой субъединице рибосомы и играет важную роль в синтезе белка. Она формирует активный сайт рибосомы, где происходит каталитическая реакция синтеза белка. Она также взаимодействует с тРНК во время транслокации, помогая перемещать тРНК и мРНК через рибосому. 23S рРНК обладает рибозимной активностью, способностью катализировать химические реакции. Она является ключевым компонентом пептидилтрансферазного центра рибосомы, который катализирует образование пептидных связей между аминокислотами в процессе синтеза белка.
3. Для выравнивания использован blastn (данный инструентр имеет общирное применение и в данно случае решать задачу некодирующих аминокислотных последовательностей белков в организмах, которые не являются близкими родственниками.(Параметры -evalue=0.05, чтобы отбросить совсем плохие находки, а -outfmt=7 для выдачи в виде таблицы)
1. blastn -task blastn -query 16rRNA.fna -db GCF_011100685.1_UU_Cfam_GSD_1.0_genomic.fna -out 16S_blm.out -outfmt 7 -evalue 0.05
Выдача blastn
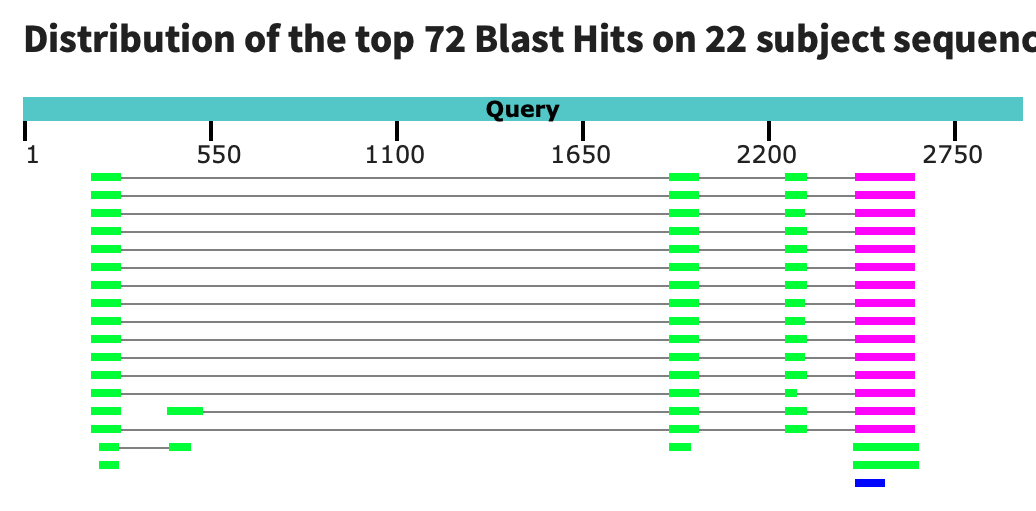
16S: 15 находок(1 хромосома(7), 14 скэффолда(unplaced))
Для лучшей визуализации принадлежность участков выравнивания (features) провела BLAST онлайн(параметры запуска)
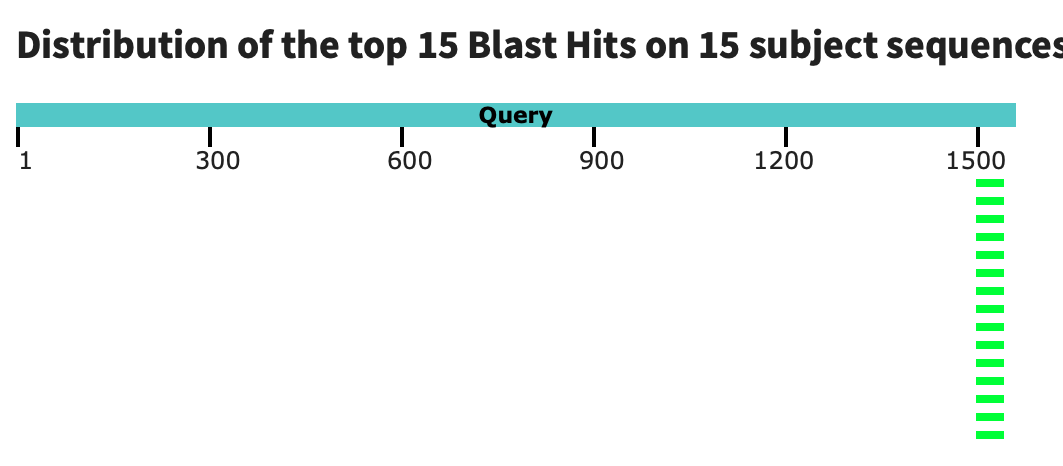
2.blastn -task blastn -query 23rRNA.fna -db GCF_011100685.1_UU_Cfam_GSD_1.0_genomic.fna -out 23S_blm.out -outfmt 7 -evalue 0.05
Выдача blastn
23S: 72 находок(15 находок по хромосомам:32(4 находки),17(5 находки),13(2 находки),29,8,2,1;unplaced genomic scaffold: 57 )