Практикум 6. Transmembrane proteins
Task 1
Omp33 porin
Type:
Transmembrane (3 classes)
Class: Beta-barrel transmembrane (36 superfamilies)
Superfamily: OmpG-like porins (n=14,S=16) (3 families) PF09381
Family: Omp33 porin (1 proteins) 1.B.1 (TCDB) PF16956 IPR031593 PDBsum
Species: Acinetobacter baumannii (37 proteins)
Localization: Bacterial Gram-negative outer membrane (421 proteins)
pdb: 6gie
Uniprot: A0A081GU02_ACIBA
Function: Omp33's main function is to serve as a channel for the passive diffusion of small molecules, such as ions and nutrients, across the bacterial outer membrane. Porins like Omp33 play a critical role in the uptake of essential nutrients and in the defense mechanisms of the bacterial cell.
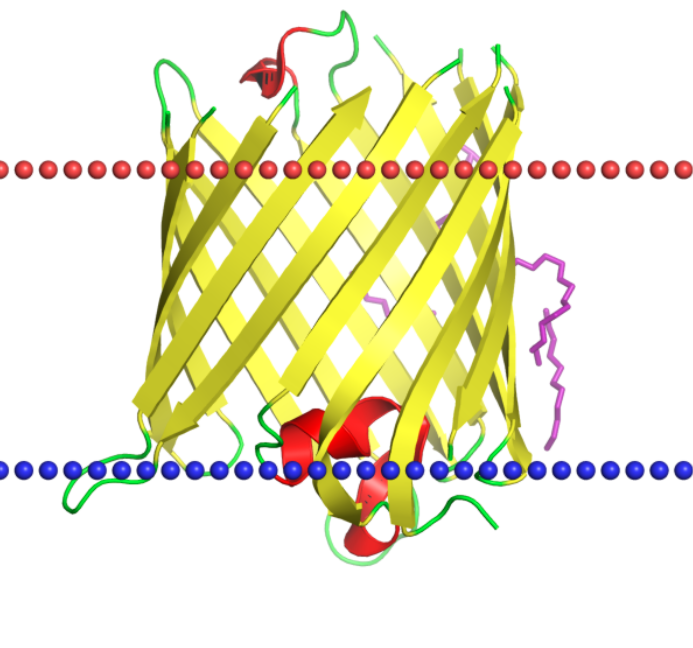
Img.1. Image of the Omp33 porin structure location in the cell membrane, this picture was taken form OPM site.
Coordinates of the transmembrane regions of the protein from OPM:
1( 3- 10)
2( 22- 30)
3( 51- 58)
4( 74- 83)
5( 90- 97)
6( 115- 121)
7( 127- 134)
8( 169- 177)
9( 185- 194)
10( 200- 206)
11( 214- 219)
12( 232- 239)
13( 246- 251)
14( 265- 272)
The protein sequence was downloaded from the PDB record in fasta format and uploaded to DeepTMHMM.
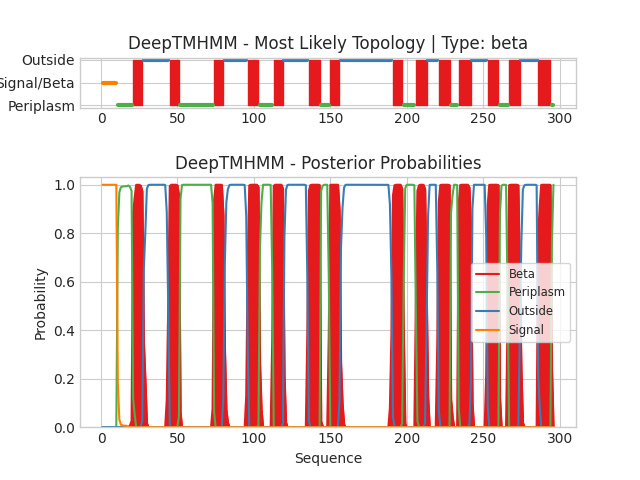
Img.2. The DeepTMHMM output
On the horizontal axes, the positions of amino acids of the protein are plotted. On the vertical axes - the probability of the predicted existence of the amino acid in a certain position. Different colors denote different positions: Red - in the membrane, green - in the periplasm,blue - outside the cell, orange - signal sequence.
The output file in .gff3results:
1(21-27)
2(45-51)
3(74-80)
4(96-103)
5(113-119)
6(136-143)
7(150-156)
8(191-197)
9(206-213)
10(221-228)
11(234-242)
12(253-260)
13(267-274)
14(286-294)
Chain Number of predicted TMRs: 14,which corresponds to the experimentally obtained model, but the boundaries of the transmembrane fragments in the experimental model and in the predicted one do not coincide at all. This could be arised from protein conformational flexibility or post-translational modifications, or even the presents of other molecules could influence localization of regions.
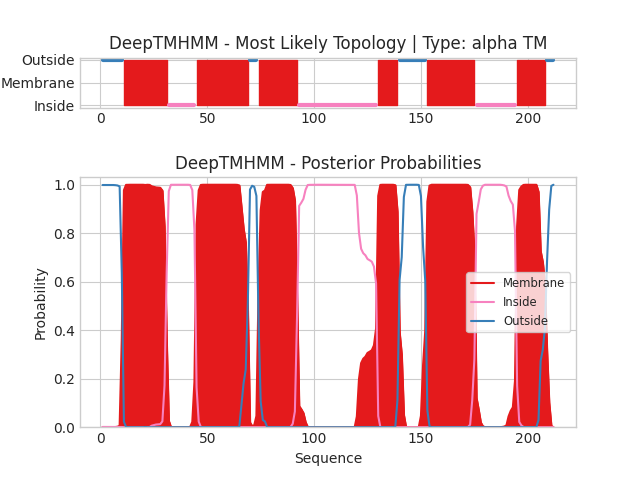
Task 2.Comparison of predictions of transmembrane regions in the alpha-helical protein
1. My protein which currently has no close homolog with an experimentally obtained three-dimensional structure (terra incognita) is Leucine efflux protein (LEUE_ECO57)
Name: Leucine efflux protein/Белок выхода лейцина
Swiss-prot AC: Q8XDS6
Organism: Escherichia coli O157:H7
Function:Exporter of leucine
2.Run DeepTMHMM with Leucine efflux protein: