EMBOSS
- Несколько файлов в формате fasta собрать в единый файл
seqret NC\*.fasta all.fasta
входные файлы
NC_010103.1.fasta
NC_010740.1.fasta
NC_010167.1.fasta
NC_012442.1.fasta
Выдача программы
seqs4.fasta
- Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы
seqretsplit seqs4.fasta seq.fasta
- Из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле
Использовали файл sequence.gb, координаты трех кодирующих последовательностей записали в файл list.txt
seqret @list.txt seqs.fastaПоследовательности записаны в файл seqs.fasta
- Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta файле.
transeq human_mito_cds.fasta trans.fasta -table 2
- Транслировать данную нуклеотидную последовательность в шести рамках.
transeq seq.fasta trans.fasta -frame 6
- Перевести выравнивание и из fasta формате в формат .msf
seqret D_AgeI.fasta msf::align.msf
-
Выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания и всеми остальными (на выходе только имя последовательности и число)
infoalign aln.msf inf.txt -refseq 2 -only -name -idcount
inf.txt
- Перевести аннотации особенностей в записи формата .gb в табличный формат .gff
featcopy NC_009894.gbk try.gff
- Из данного файла с хромосомой в формате .gb получить fasta файл с кодирующими последовательностями; (*) добавить в описание каждой последовательности функцию белка (из поля product)
extractfeat NC_009894.gbk info.fasta -type CDS -describe product
- Перемешать буквы в данной нуклеотидной последовательности;
shuffle -o shuf.fasta NC_009894.gbk
- Найдите частоты кодонов в данных кодирующих последовательностях
cusp human_mito_cds.fasta table.txt
table.txt
- Найдите частоты динуклеотидов в данной нуклеотидной последовательности и сравните их с ожидаемыми
compseq human_mito_cds.fasta -word 2 info.txt -calcfreq в файле info.txt есть таблица в которой в 3 колонке найденная частота в 4ой колонке ожидаемая частота, в 5ой колонкe отношение вычисленной частоты к ожидаемой частоте
- Выровняйте кодирующие последовательности соответственно выравниванию белков - их продуктов
tranalign cdss_D-ddeI.fasta proteins_D-DdeI.fasta align.fasta
Карта локального сходства геномов.
Мы построили карту локального сходства геномов бактерий Thermotoga maritima MSB8 NC_023151.1 и Thermotoga neapolitana DSM 4359 NC_011978.1
Для построения карты локального сходства использовался blast2seq (алгоритм blastn) на сайте NCBI. Характеристики полученного выравнивания приведены в таблице 1.
ВыравниваниеТаблица 1. Характеристики выравнивания геномов
| e-value | 0.0 |
|---|---|
| identity | 97% |
| query cover | 87% |
| max score | 1.638e+05 |
| total score | 1.374e+06 |
Рис.1 Карта локального сходства
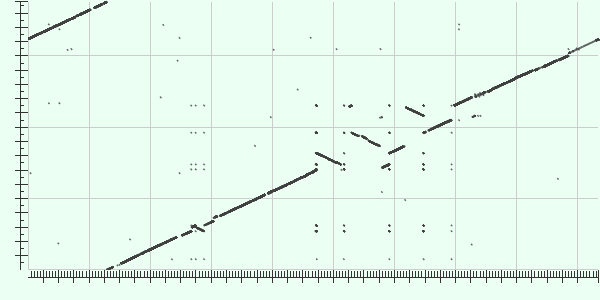
Рассмотрим по данной карте крупные эволюционные события произошедшие с геномами на пути от общего предка.
Анализ карты | |
Изображение | Описание |
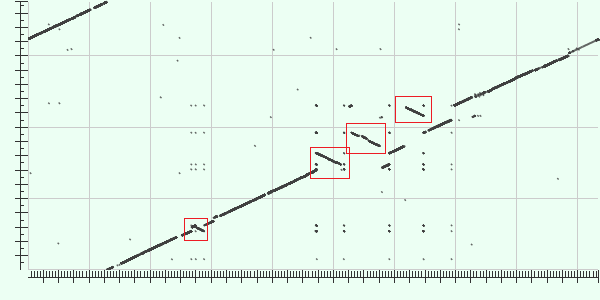 |
Красным выделены инверсии. |
 |
Синим выделены участоки, на котором можно наблюдать индель. (А) Произошла вставка в геноме T.maritima или делеция в геноме T.neapolitana (В) Аналогично. |
| |
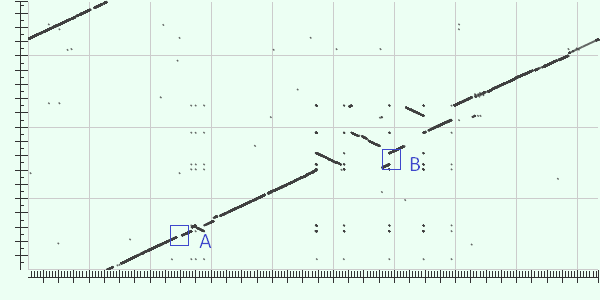
Произошла транслокация: участки А и В поменялись местами в геноме T.maritima и в геноме T.neapolitana. Также произошла инверсия участка В. |
| |
Участки С и D также поменялись местами в двух геномах. Причем на участке D, видимо произошла вставка или делеция. |
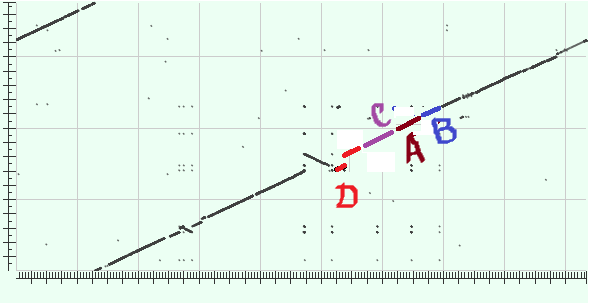 |
Т.е. эти участки могли бы располагаться примерно так. |
| |
И, наверное, на участке D не индель, а снова транслокация. Или участок Е -- это и есть вставка |
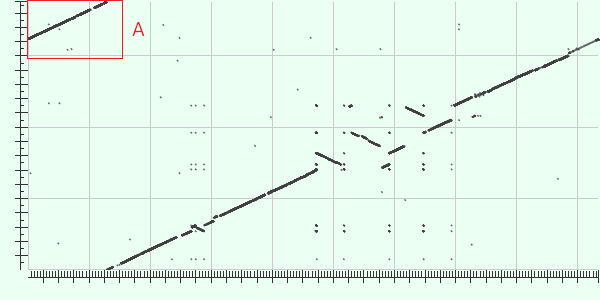 |
У рассматриваемых бактерий кольцевые хромосомы. Из-за того, что по-разному установлены координаты, возникает участок, выделенный рамкой. |