Для выполнения задания использовался код, написанный моей однокурсницей, Еленой Гончаровой.
Выравнивания, полученные разными программами: MUSCLE, MAFFT, T-COFFEE.
Ссылка на проект JalView
Были получены следующие результаты:
| № | MUSCLE | MAFFT | Длина |
|---|---|---|---|
| 1 | (17,39) | (17,39) | 23 |
| 2 | (95,98) | (95,98) | 4 |
| 3 | (63,65) | (64,66) | 3 |
| 4 | (59,60) | (60,61) | 2 |
| № | MUSCLE | MAFFT | Длина |
|---|---|---|---|
| 1 | (40,58) | (40,58) | 19 |
| 2 | (1,16) | (1,16) | 16 |
| 3 | (61,62) | (62,63) | 3 |
Одинаково выровненные колонки, не входящие в блоки: (41,41)
Процент совпадающих колонок в MUSCLE: 33,7%
| № | MUSCLE | T-COFFEE | Длина |
|---|---|---|---|
| 1 | (15,39) | (19,43) | 25 |
| 2 | (62,65) | (73,76) | 4 |
| 3 | (57,58) | (68,69) | 2 |
| № | MUSCLE | T-COFFEE | Длина |
|---|---|---|---|
| 1 | (40,60) | (40,60) | 21 |
| 2 | (1,14) | (1,14) | 14 |
Одинаково выровненные колонки, не входящие в блоки: (41,45), (60,71), (94,108), (97,112).
Процент совпадающих колонок в MUSCLE: 35,7%
Количество совпадающих колонок при выравнивании с помощью программ MUSCLE и T-COFFEE незначительно больше, чем с помощью MUSCLE и MAFFT. Однако при выравнивании с помощью MUSCLE и MAFFT совпадающие колонки имеют примерно одинаковые координаты, что говорит о большей схожести этих алгоритмов.
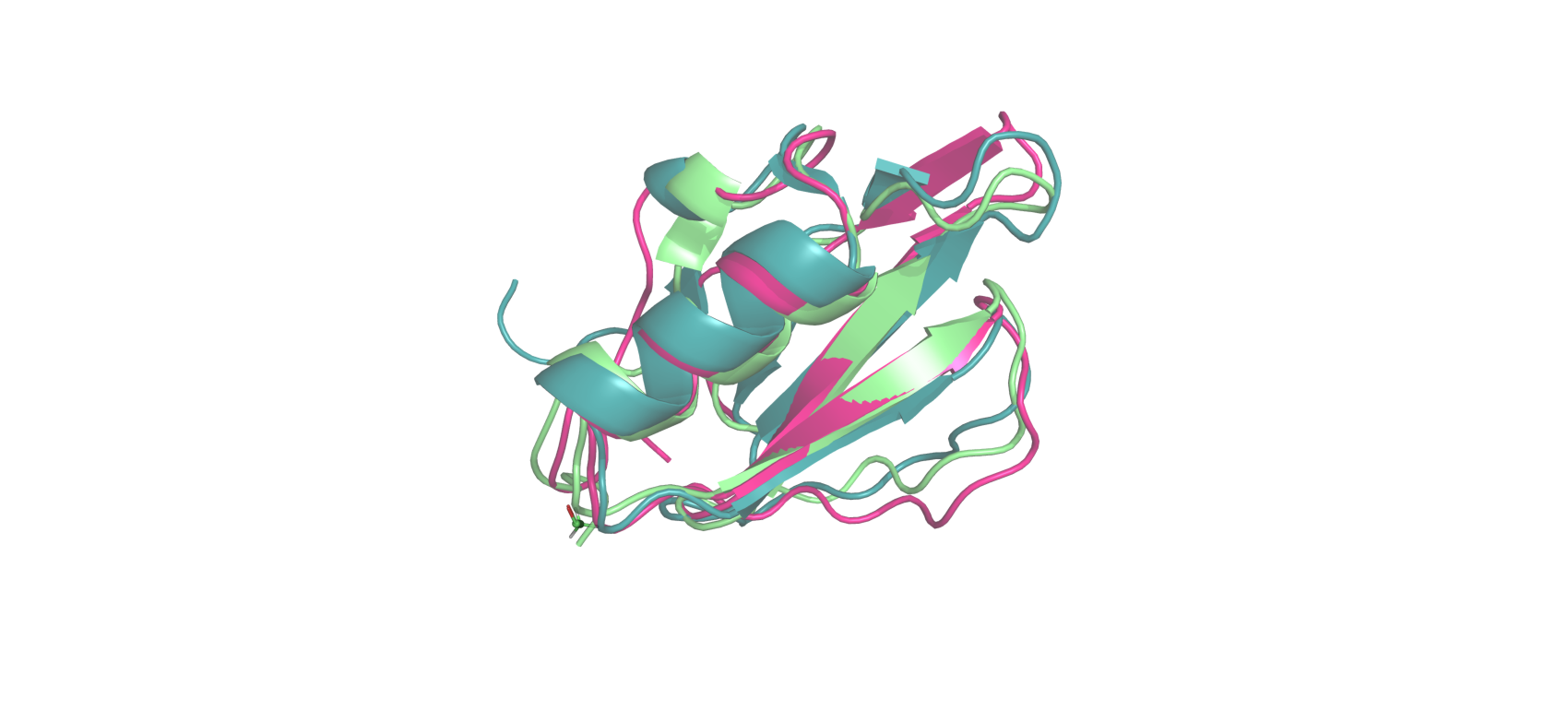
Были выбраны 3 белка: 1DWM, 1EGL, 1MIT. С помощью PDBeFold были получены структурные выравнивания. Также последовательности были выровнены с помощью MUSCLE. Полученные выравнивания были проанализированы скриптом из задания 2.
Были получены следующие результаты:
| № | MUSCLE | PDBeFold | Длина |
|---|---|---|---|
| 1 | (1,41) | (1,41) | 41 |
| 2 | (46,58) | (47,59) | 13 |
| 3 | (68,70) | (71,73) | 3 |
Одиночных совпадений вне блоков: 0.
Несовпадающие участки в 12.3_pdb.fa:
42-45 (длина 4)
59-67 (длина 9).
Процент совпадения: 81,4%.
Из полученных результатов можно сделать вывод, что последовательности обладают высоким сходством, оба алгоритма выравнивания справляются со своей задачей, выравнивание, полученное из структурного, является правдивым и точным.
MAFFT (Multiple Alignment using Fast Fourier Transform) — это высокопроизводительная программа для множественного выравнивания биологических последовательностей (белков, ДНК, РНК), разработанная группой Кадзутаки Като [1]. Впервые программа была выпущена в 2002 году [1] и с тех пор активно развивается (текущая стабильная версия — 7) [3,4].
Применение
MAFFT широко используется в биоинформатике для:
Ограничения
Как и любые автоматические методы, MAFFT может ошибаться при выравнивании сильно дивергировавших последовательностей с большим количеством вставок/делеций [3]. Разработчики рекомендуют визуально проверять и при необходимости корректировать результаты.
Список литературы
[1] Katoh K. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform // Nucleic Acids Research. 2002. T. 30. № 14. С. 3059–3066
DOI: 10.1093/nar/gkf436
[2] Katoh K. MAFFT version 5: improvement in accuracy of multiple sequence alignment // Nucleic Acids Research. 2005. T. 33. № 2. С. 511–518
DOI: 10.1093/nar/gki198
[3] Katoh K., Standley D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability // Molecular Biology and Evolution. 2013. T. 30. № 4. С. 772–780
DOI: 10.1093/molbev/mst010
[4] Katoh K., Rozewicki J., Yamada K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization // Briefings in Bioinformatics. 2017. T. 20. № 4. С. 1160–1166
DOI: 10.1093/bib/bbx108
[5] Long H., Li M., Fu H. Determination of optimal parameters of MAFFT program based on BAliBASE3.0 database // SpringerPlus. 2016. T. 5. № 1
DOI: 10.1186/s40064-016-2526-5
[6] Nakamura T. и др. Parallelization of MAFFT for large-scale multiple sequence alignments // Bioinformatics. 2018. T. 34. № 14. С. 2490–2492
DOI: 10.1093/bioinformatics/bty121
[7] Zheng W. и др. A Modified Multiple Alignment Fast Fourier Transform with Higher Efficiency // IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2017. T. 14. № 3. С. 634–645
DOI: 10.1109/tcbb.2016.2530064