Сравние выравнивания одних и тех же последовательностей разными программами
Для выполнения работы был выбран белковый домен Arrestin (or S-antigen), N-terminal domain, AC - PF00339 из практикума 11.
Выравнивание проводили с помощью программ: MAFFT, MUSCLE, T-COFFEE. Ниже предоставлен файл Jalview cо сравниваемыми выравниваниями одних и тех же последовательностей
Далее мы сравнили полученные результаты, используя программу Macho.py , сделанную однокурсниками, получили список одинаково выровненных колонок и представили их в таблице 1.
Видно, что программы Mafft t-coffee имеют большую схожесть, чем mafft, muscle. Разница в результатах выравнивания между программами заключается в разных алгоритмических подходах. Несмотря на то, что все три программы используют прогрессивный и итеративный метод, Muscle принципиально отличается от T-coffee тем, что она использует метод, который предполагает, что мутации происходят с постоянной скоростью, в то время как T-coffee - что скорость мутаций непостоянна. Для выполнения задания выбрали 3 белка семейства Arrestin из Pfam:
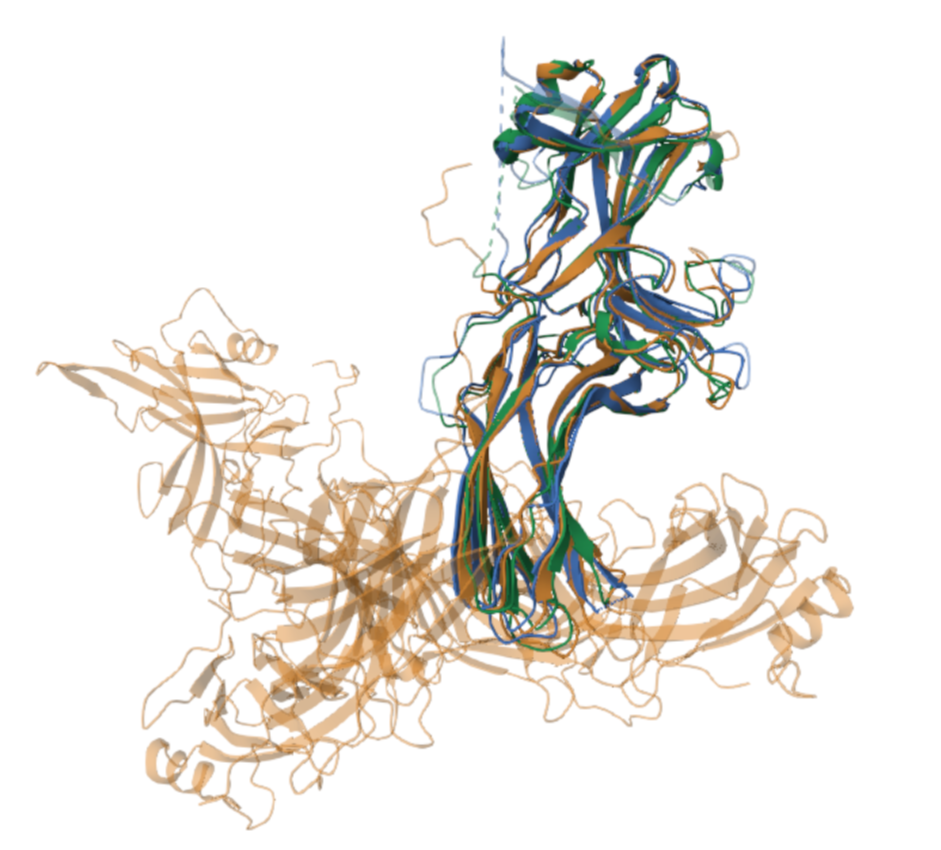
1) ARRS_BOVIN (1ayr). Белок связывается с фосфорилированным, активированным светом родопсином (RHO) и прерывает передачу сигналов RHO через G-белки. 2) ARRB1_BOVIN (1g4m). Участвует в регуляции передачи сигналов через рецепторы, связанные с G-белками (GPCR) и действует как "выключатель" для рецепторов, чтобы клетка не перегружалась сигналами. 3) ARRB2_BOVIN (3p2d). Функционально схож с ARRB1_BOVIN, но он работает с широким спектром GPCR (дофамин, адренорецепторы) и связывает рецепторы медленнее, но регулирует больше сигнальных путей. Далее было проведено два выравнивания разными способами: программой MSA и из совмещения структур. В проекте Jalview представлены 2 выравнивания, которые мы сравним, используя программу Macho.py. На выходе получили список одинаково выровненных колонок у выбранных последовательностей. Результаты приведены в таблице 2. Длина структурного выравнивания: 385 Длина выравнивания Mafft: 374 Процент совпадающих колонок в первом выравнении (Mafft): 91.18% Процент совпадающих колонок во втором выравнении (PDB): 88.57% Видно, что выравнивания очень похожи, но не идентичны, самый большой консервативный блок одинаково выровненных колонок (200-337)-(205-342). Так как 2 выравнивания имеет высокий процент совпадающих колонок, то можно сделать вывод о том, что выравнивание достаточно близко к эволюционному. Для наглядности добавим рисунок 1 - визуальное представление совмещения трех структур. Визуальный анализ показал высокую степень совпадения структурных элементов, что подтверждает консервативность доменной организации белков семейства Arrestin. MUSCLE (от англ. Multiple Sequence Comparison by Log-Expectation) — компьютерная программа для выравнивания множественных последовательностей белковых и нуклеотидных последовательностей. Программа была разработан Робертом Эдгаром (Robert C. Edgar) и впервые представлен в 2004 году. Элементы алгоритма включают быструю оценку расстояния с помощью подсчёта k-меров, прогрессивное выравнивание с помощью новой функции профиля, которую мы называем оценкой логарифмического ожидания, и уточнение с помощью зависимого от дерева ограниченного разбиени[1] . Ключевые особенности:
- Высокая скорость: Оптимизированный алгоритм позволяет обрабатывать большие наборы данных (до ~10 000 последовательностей). - Трехэтапный алгоритм: 1)Грубое выравнивание 2)Уточнение дерева 3)Итеративное улучшение - Поддержка разных форматов: FASTA, CLUSTAL, MSF и др. - В 2-10 раз быстрее ClustalW. - Эффективен для умеренно гомологичных последовательностей. - Менее точен для сильно дивергентных последовательностей. - Ограниченная поддержка структурной информации [2]
Mafft и Muscle
Mafft и T-coffee
Блоки одинаково выровненных колонок
(29-40)-(18-29)
(49-56)-(38-45)
(142-15)-(115-127)
(157-186)-(130-159) (15-16)-(18-19)
(20-21)-(10-11)
(31-39)-(23-31)
(49-61)-(43-55)
(113-115)-(96-98)
(141-186)-(127-172)
(201-202)-(186-187)
(214-216)-(205-207)
Одинаково выровненные колонки, не входящие в блоки
(105)-(87)
(191)-(177)
(195-183) Совмещение структур и сравнение с программой MSA
Таблица 13-1
Mafft и PDB
Блоки одинаково выровненных колонок
(1-94)-(1-94)
(99-157)-(100-158)
(159-161)-(161-163)
(168-187)-(171-190)
(200-337)-(205-342)
(340-343)-(345-348)
(346-362)-(351-367)
(364-368)-(369-373)
Одинаково выровненные колонки, не входящие в блоки
(374)-(385)
Программа MSA - Muscle