Практическая работа №14: Сборка генома de novo
1. Подготовка чтений программой Trimmomatic.
В работе использовались одиночные чтения, полученные в результате секвенирования бактерии Buchnera aphidicola str. Tuc7. Код доступа: SRR4240378. Данные были скачены следующей командой:
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/008/SRR4240378/SRR4240378.fastq.gzПоскольку чтения могут содержать остатки адаптерных последовательностей, их необходимо удалить. Для этого использовалась программа Trimmomatic. Все известные адаптерные последовательности Illumina были объединены в файл adapters.fasta (54 последовательности).
Команда для объединения: code>cat /mnt/scratch/NGS/adapters/*.fa > adapters.fastaОчистка проводилась в два этапа:
1. Удаление адаптеров:
TrimmomaticSE SRR4240378.fastq.gz SRR4240378_trim.fastq.gz ILLUMINACLIP:adapters.fasta:2:7:7Вывод программы:
Input Reads: 4420587 Surviving: 4338744 (98.15%) Dropped: 81843 (1.85%) TrimmomaticSE: Completed successfully
Адаптерами оказалось 1.85% чтений.
2. Фильтрация по качеству и длине:
TrimmomaticSE SRR4240378_trim.fastq.gz SRR4240378_trimf.fastq.gz TRAILING:20 MINLEN:32TRAILING:20 — удалит нуклеотиды с правого конца, если их качество < 20
MINLEN:32 — удалит чтения короче 32 нуклеотидов
Вывод программы:
Input Reads: 4338744 Surviving: 4154738 (95.76%) Dropped: 184006 (4.24%) TrimmomaticSE: Completed successfully
Удалено 184 006 чтений (4.24%).
Результаты очистки
| Этап | Число чтений | Процент сохраненных | Удалено |
|---|---|---|---|
| Исходные данные | 4 420 587 | 100% | — |
| После удаления адаптеров | 4 338 744 | 98.15% | 81 843 (1.85%) |
| После фильтрации качества | 4 154 738 | 95.76% | 184 006 (4.24%) |
2. Сборка генома программой Velvet
Дальше запустили Velvet — сборщик геномов. Velvet находит перекрытия между чтениями и склеивает их в контиги, используя алгоритм графа де Брёйна. Работает в 2 этапа:
Этап 1. Подготовка k-меров:
Все исходные чтения разбиваются на короткие перекрывающиеся фрагменты фиксированной длины (k-меры), в нашем случае их длина = 31.
Команда
velveth Assem 31 -fastq -short ../SRR4240378_trimf.fastq Этап 2. Сборка контигов:
velveth Assem 31 -fastq -short SRR4240378_trimf.fastqСозданы файлы: contigs.fa, Graph, LastGraph, Log, PreGraph, Roadmaps, Sequences, stats.txt.
Итог сборки:
N50 - 7028 bp Максимальная длина контига - 36 746 bp Общее количество нуклеотидов - 657 295 bp Число контигов - 3693. Анализ контигов
Для дальнейшего анализа были выбраны три наиболее длинных контига:
grep ">" Assem/contigs.fa | sort -t_ -k4,4nr | head -3Объяснение параметров: grep ">" — ищет все строки с описанием контигов sort -t_ — разделитель полей нижнее подчёркивание _ -k4,4nr — сортировка по 4-му полю (длина) в обратном порядке head -3 — покажет топ 3
>NODE_8_length_36746_cov_20.017199 >NODE_57_length_19371_cov_20.546642 >NODE_15_length_16745_cov_20.901762
| Контиг | Длина (bp) | Покрытие |
|---|---|---|
| NODE_8 | 36 746 | 20.02 |
| NODE_57 | 19 371 | 20.55 |
| NODE_15 | 16 745 | 20.90 |
Аномалии покрытия
Среднее покрытие по всем контигам составляет 16.9 ( Использовали команду: grep ">" Assem/contigs.fa | awk -F'_' '{sum+=$6; count++} END{print "Среднее покрытие:", sum/count}'). Контиги, покрытие которых более чем в 5 раз отличается от этого значения, могут указывать на возможные ошибки сборки или биологичнские особенности.
Расчитаем пороги для аномалий (отличаются от медианы более чем в 5 раз):
- Нижний порог: 16.9 / 5 ≈ 3.4
- Верхний порог: 16.9 × 5 ≈ 84.5
Контиги с аномально высоким покрытием (>84.5×):
grep ">" Assem/contigs.fa | awk -F'_' '$6 > 84.5' | head -5Контиги с аномально низким покрытием (<3.4×):
grep ">" Assem/contigs.fa | awk -F'_' '$6 < 3.4' | head -5Oбнаружены следующие аномалии:
1. Контиги с аномально высоким покрытием: NODE_19: длина 2,106 bp, покрытие 100.56× (в 5.95 раз выше среднего 16.9×). Возможно, представляет повторяющийся регион (например, рибосомные гены). NODE_81: длина 934 bp, покрытие 102.75× (в 6.08 раз выше среднего).
2. Контиги с аномально низким покрытием: NODE_166: длина 64 bp, покрытие 2.84× (в 5.95 раз ниже среднего). Вероятно, ошибка сборки или загрязнение. NODE_271: длина 31 bp, покрытие 3.23× (в 5.23 раз ниже среднего). NODE_281: длина 72 bp, покрытие 2.94× (в 5.75 раз ниже среднего).
4. Сравнительный анализ с референсным геномом
Для сравнения был выбран полный геном Buchnera aphidicola штамма из тли Schizaphis graminum (accession: NZ_CP029205.1), длина: 630 854 bp.
Каждый из трёх самых длинных контигов был сравнен с референсным геномом с помощью программы Megablast.
1. Первый контиг: NODE_8_length_36746_cov_20.017199 (рис.1)
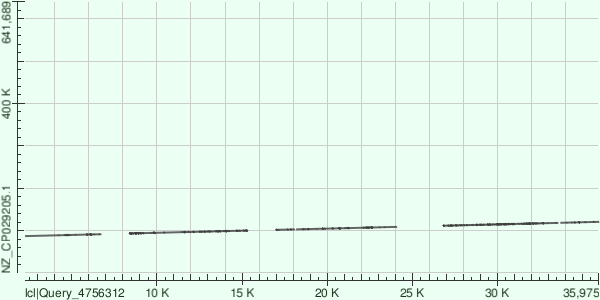
Координаты участка хромосомы: Sbjct: 87013–91478. Соответствующий участок контига располагается в пределах: Query: 2248–6761
Характеристики выравнивания
- Длина выравнивания: 4514 п.н.
- Число совпадающих нуклеотидов: 3722 (≈82%)
- Число однонуклеотидных различий: 792
- Число гэпов: 72 (≈1%)
- E-value: 0.0
- Покрытие контига выравниванием 74%
В результате BLAST-поиска был обнаружен один значимый хит, соответствующий хромосоме референсного генома. Выравнивание характеризуется нулевым E-value, что указывает на его высокую статистическую значимость. На графике видно, что контиг выравнивается с хромосомой несколькими фрагментами, которые имеют одинаковую ориентацию и располагаются последовательно вдоль одного участка хромосомы. Это указывает на то, что данный контиг действительно соответствует участку бактериального генома.
2. Второй контиг: NODE_57_length_19371_cov_20.546642 (рис.2)
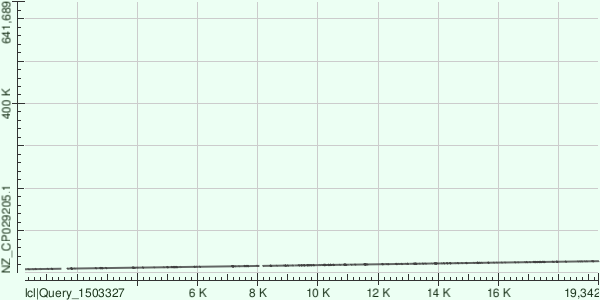
Координаты участка хромосомы: Sbjct: 16750–27901.х Соответствующий участок контига располагается в пределах: Query: 8183–19577
Характеристики выравнивания
- Длина выравнивания: 11 395 п.н.
- Число совпадающих нуклеотидов: 8 676 (≈76%)
- Число однонуклеотидных различий: 2 719
- Число гэпов: 478 (≈4%)
- E-value: 0.0
- Покрытие контига выравниванием 97%
График локального сходства демонстрирует непрерывную ровную линию, что указывает на соответствие исследуемого контига одному протяжённому участку хромосомы выбранной бактерии и подтверждает их гомологию.
3. Третий контиг: NODE_15_length_16745_cov_20.901762 (рис.3)
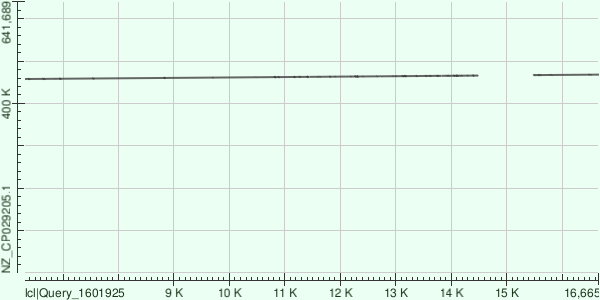
Координаты участка хромосомы: Sbjct: 457626–465787.х Соответствующий участок контига располагается в пределах: Query: 6315–14573
Характеристики выравнивания
- Длина выравнивания: 8 259 п.н.
- Число совпадающих нуклеотидов: 6 540 (≈79%)
- Число однонуклеотидных различий: 1 719
- Число гэпов: 190 (≈2%)
- E-value: 0.0
- Покрытие контига выравниванием 56%
Неполное покрытие (56%) может объясняться различиями между анализируемым штаммом и выбранным референсом, а также особенностями de novo сборки (короткие чтения, возможные разрывы/сложно собираемые участки). При этом достаточно высокий процент идентичности в выровненных частях показывает, что совпавшие фрагменты соответствуют бактериальному геному и не являются случайными.