Предсказание вторичной структуры заданной тРНК
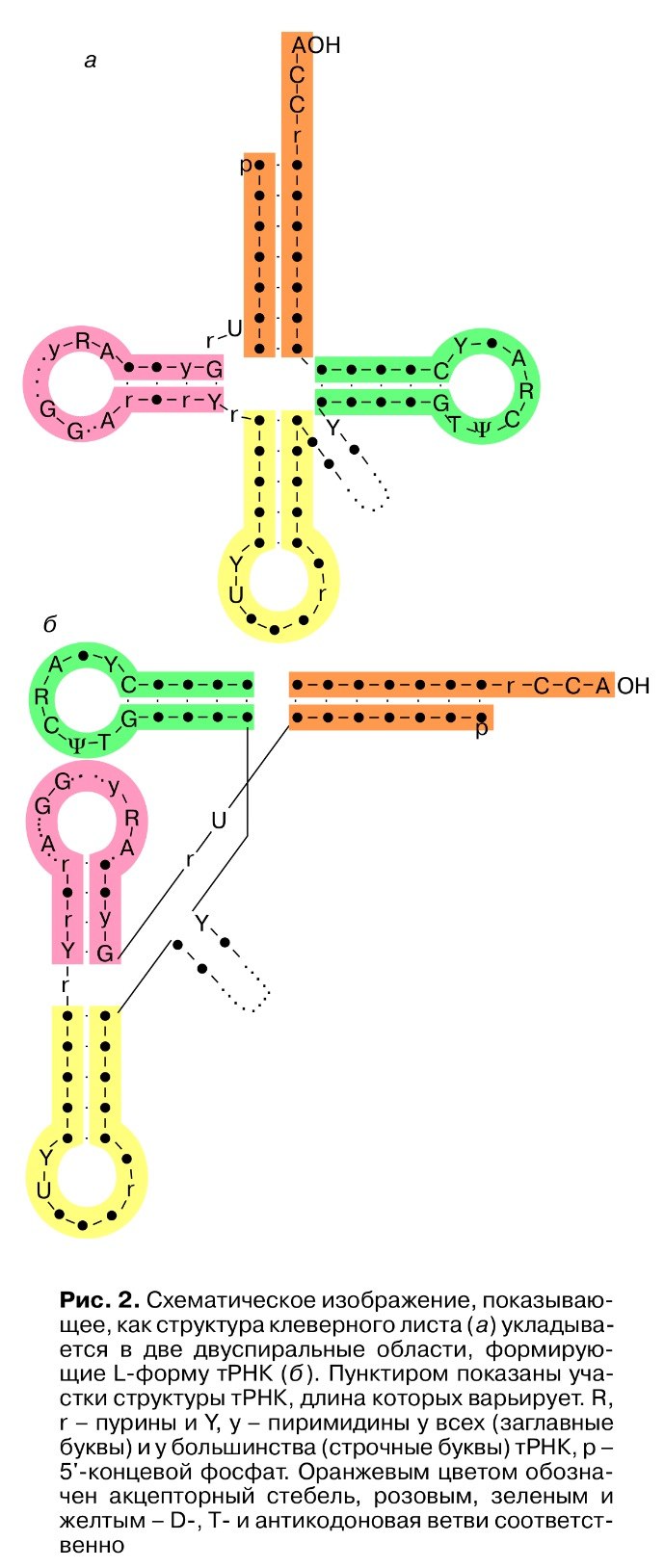 В этом задании необходимо было сравнить реальную и предсказанные вторичные структуры заданной
тРНК (PDB ID 1I9V), полученные с помощью find_pair (реальная структура) и двух методов предсказания - путем
поиска инвертированных повторов и по алгоритму Зукера. Схематическое изображение участков
вторичной структуры показано на рисунке 2 [1].
В этом задании необходимо было сравнить реальную и предсказанные вторичные структуры заданной
тРНК (PDB ID 1I9V), полученные с помощью find_pair (реальная структура) и двух методов предсказания - путем
поиска инвертированных повторов и по алгоритму Зукера. Схематическое изображение участков
вторичной структуры показано на рисунке 2 [1].
Поиск инвертированных повторов проводился с помощью программы einverted из пакета EMBOSS,
которая получает на вход последовательность нуклеотидов, а на выходе дает файлы sequence.fasta и
sequence.inv, содержащие информацию об обнаруженных комплементарных участках
последовательности и предполагаемых водородных связях на этих участках соответственно.
Когда я использовала программу с параметрами по умолчанию, файл sequence.fasta оказывался пустым,
поэтому для корректной работы пришлось подбирать параметры вручную. Наиболее
значимые изменения вызывало варьирование параметра Minimum score threshold, который по умолчанию был
равен 50. Программа начинала выдавать результат при понижении threshold до 16, однако наиболее близкие
к реальной структуре предсказания были получены при threshold 14 и ниже. Наилучший результат, которого
мне удалось добиться с помощью этой программы - полное предсказание для одного из стеблей и частичное,
с несовпадением конца и начала, для другого.
Далее была использована программа RNAfold из пакета Viena Rna Package, которая
реализует алгоритм Зукера. RNAfold принимает на вход последовательность РНК и
рассчитывает вторичную структуру РНК с минимальной свободной энергией (mfe) в формате DBN
(Dot-Bracket Notation). Точки обозначают неспаренные нуклеотиды, круглые скобки - спаренные.
Квадратные и фигурные скобки обозначают взаимодействия, формирующие псевдоузлы.
Для моей структуры RNAfold сработала достаточно хорошо, причем с первого раза. Были абсолютно правильно
предсказаны 3 из 4 стеблей и один из стеблей был предсказан со сдвигом на один нуклеотид.
| Реальная и предсказанная вторичные структуры тРНК из файла 1i9v.pdb | |||
|---|---|---|---|
| Участок структуры | Позиции в структуре (результаты find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 5'-1-7-3' 5'-66-72-3' всего 7 пар |
предсказано 0 пар из 7 реальных | предсказано 7 пар из 7 реальных, стебель предсказан полностью |
| D-стебель | 5'-10-13-3' 5'-22-25-3' всего 4 пары |
предсказано 0 пар из 4 реальных | предсказано 4 пары из 4 реальных, стебель предсказан полностью |
| T-стебель | 5'-49-53-3' 5'-61-65-3' всего 5 пар |
предсказано 5 пар из 5 реальных, стебель предсказан полностью, начало и конец совпадают | предсказано 4 пары из 5 реальных + одна лишняя пара, начало и конец стебля не совпадают |
| Антикодоновый стебель | 5'-40-44-3' 5'-26-30-3' всего 5 пар |
предсказано 5 пар из 5 реальных, но начала и концы стеблей в реальной и предсказанной структурах не совпадают, предсказано 5 лишних пар | предсказано 5 пар из 5 реальных, стебель предсказан полностью |
| Общее число канонических пар нуклеотидов | 23 | 13 | 23 |
| Точечный график, показывающий вероятность образования пар оснований, RNAfold | Предсказанная вторичная структура тРНК с минимальной свободной энергией, RNAfold |
|---|---|
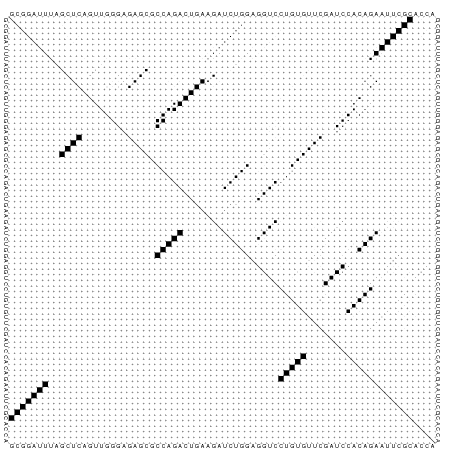 |
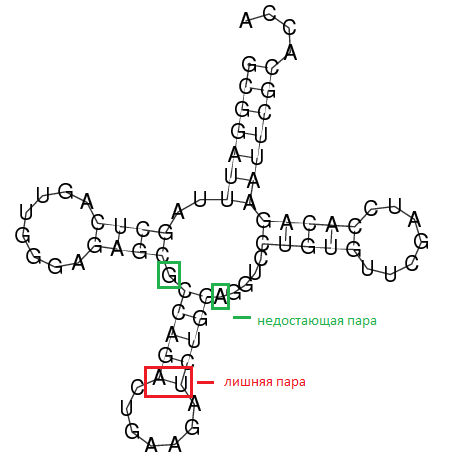 |
Поиск ДНК-белковых контактов в структуре PDB ID: 1MDM
При нажатии на окно апплета скрипт запустится автоматически. Для продолжения работы скрипта после паузы используйте кнопку resume.
Текст скрипта
Результаты работы представлены в таблице.
| Контакты атомов белка с: | Полярные | Неполярные | Всего |
|---|---|---|---|
| остатками 2'-дезоксирибозы | 7 | 51 | 58 |
| остатками фосфорной кислоты | 17 | 16 | 33 |
| остатками азотистых оснований со стороны большой бороздки | 3 | 16 | 19 |
| остатками азотистых оснований со стороны малой бороздки | 4 | 9 | 13 |
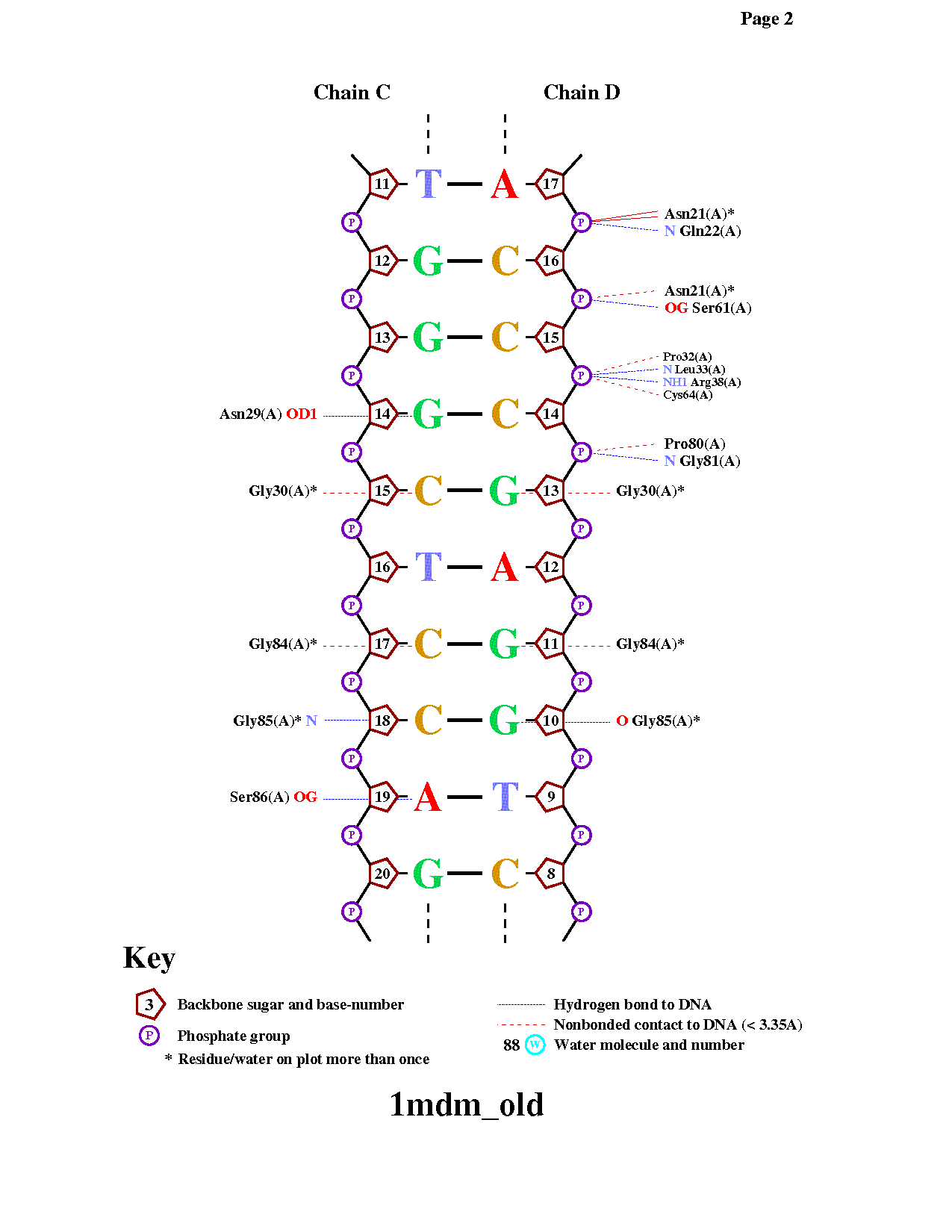
| Cхема ДНК-белковых контактов в комплексе 1MDM |
|---|
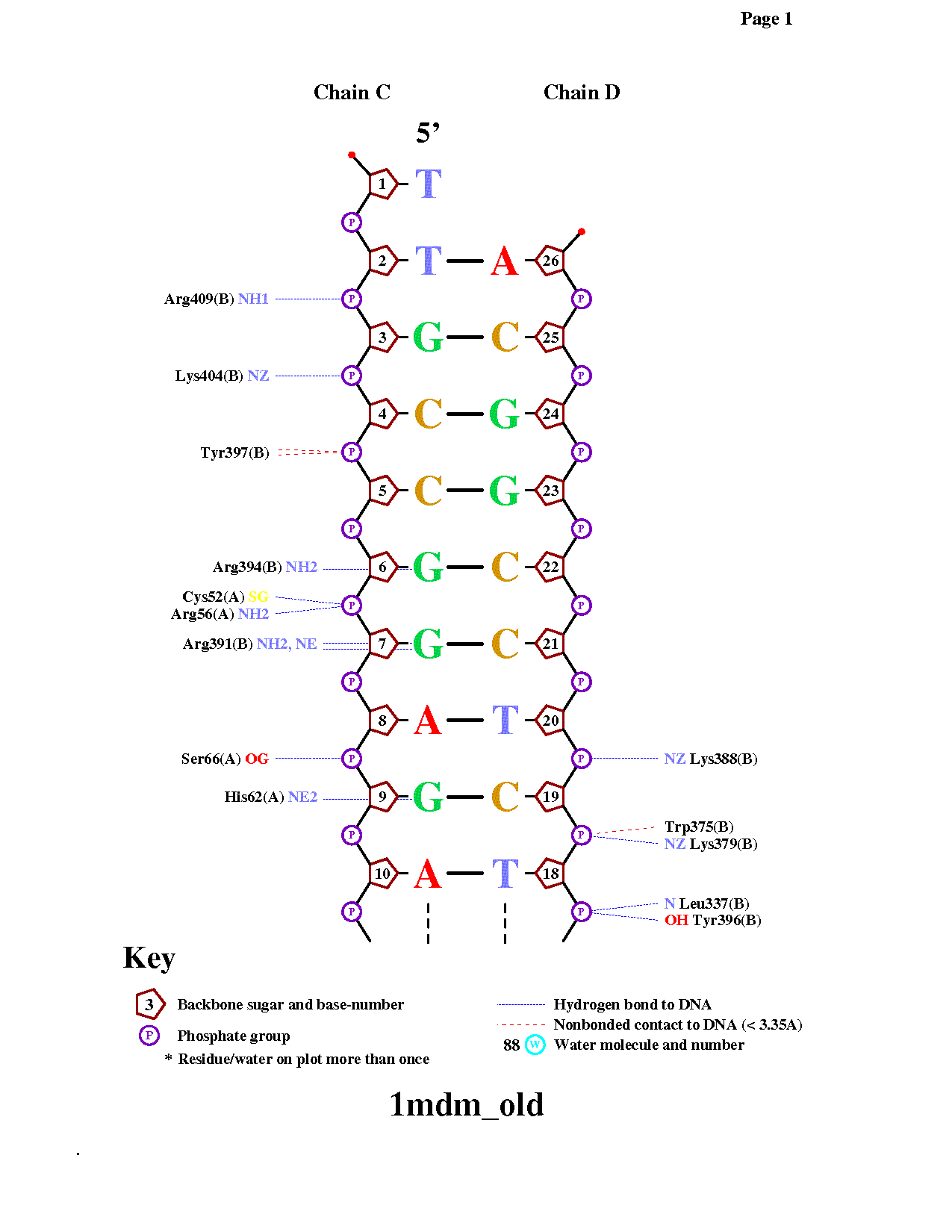 |
 |
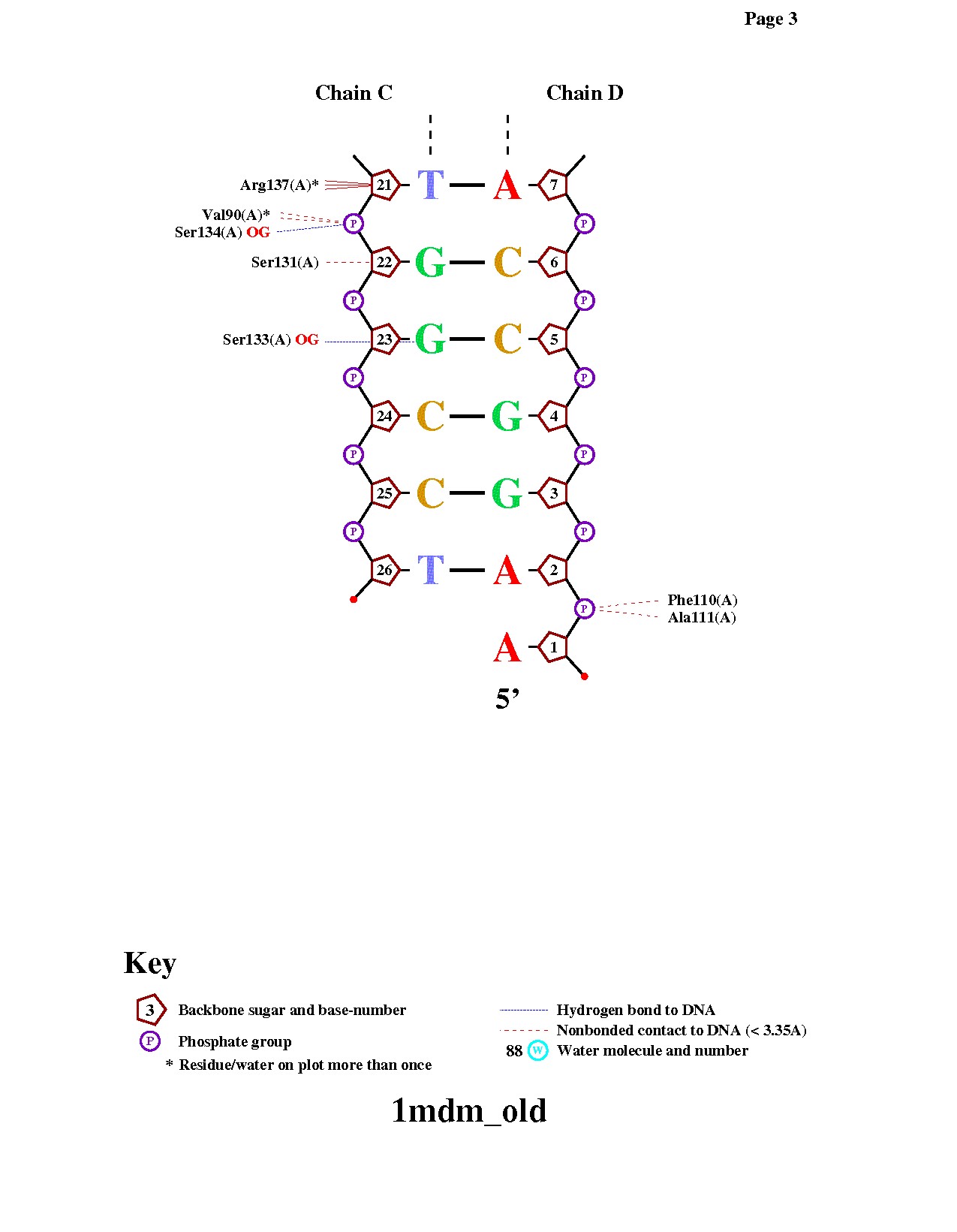 |
Далее было необходимо получить популярную схему ДНК-белковых контактов в комплексе 1MDM с помощью программы nucplot. Так как она не работает с новыми pdb, для начала нужно было воспользоваться программой remediator, чтобы получить файл в старом pdb-формате.
Посредством команды nucplot 1CF7_old.pdb был получен файл nucplot.ps, который затем был конвертирован в изображение.
Аминокислотные остатки с наибольшим числом указанных на схеме контактов с ДНК - Asn21(A) (2 контакта с одним остатком фосфорной кислоты и 1 с другим) и Arg137(A) (3 контакта с одним остатком фосфорной кислоты).
На мой взгляд, для распознавания последовательности ДНК наиболее важны аминокислотные остатки, контактирующие с остатками азотистых оснований, а не с остовом, так как именно они могут обеспечить специфичное связывание и помочь в определении последовательности. Соответсвенно, в моем комплексе наиболее важными я посчитала аминокислотные остатки, образующие наибольшее число таких контактов - Gly84(A) и Gly30(A) (см. page 2). Каждый из них образует по 2 контакта с комплементарными C и G.
C помощью JMol были получены изображения, иллюстрирующие контакты выбранных аминокислотных остатков с ДНК. Контактирующие аминокислотные остатки и остатки азотистых оснований изображены в виде wireframe c раскраской cpk.
| Аминокислотные остатки с наибольшим числом контактов с ДНК в комплексе 1MDM |
|---|
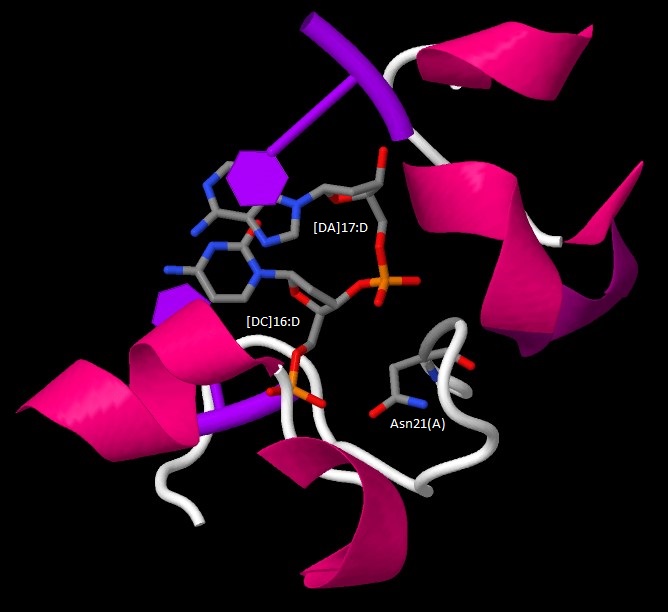 |
| Контакт Asn21(A) с остатками фосфорной кислоты [DC]16:D и [DA]17:D |
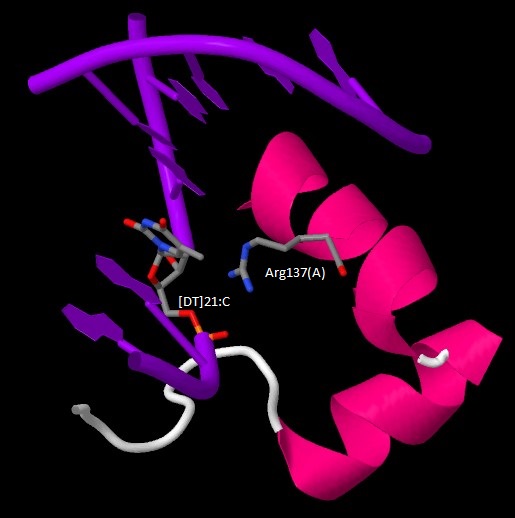 |
| Контакт Arg137(A) с остатками фосфорной кислоты [DT]21:C |
| Аминокислотные остатки, наиболее важные для распознавания последовательности ДНК в комплексе 1MDM | |
|---|---|
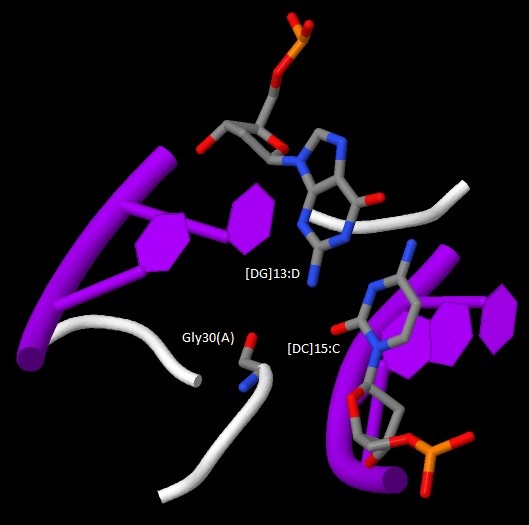 | 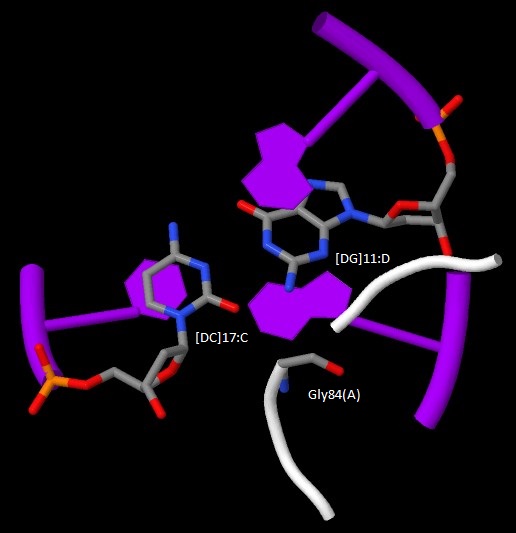 |
| Контакт Gly30(A) c остаками азотистых оснований [DG]13:D и [DC]15:C | Контакт Gly84(A) c остаками азотистых оснований [DG]11:D и [DC]17:C |