Нуклеотидный BLAST
Таксономия и функции прочтенной последовательности
Для определения таксономии использовалась последовательность final.fasta, прочтенная в практикуме
6. Был запущен нуклеотидный BLAST с алгоритмом blastn (Somewhat similar sequences) по базе данных Nucleotide collection (nr/nt)
при дефолтных параметрах.
Результат работы blastn можно увидеть на рисунке ниже.
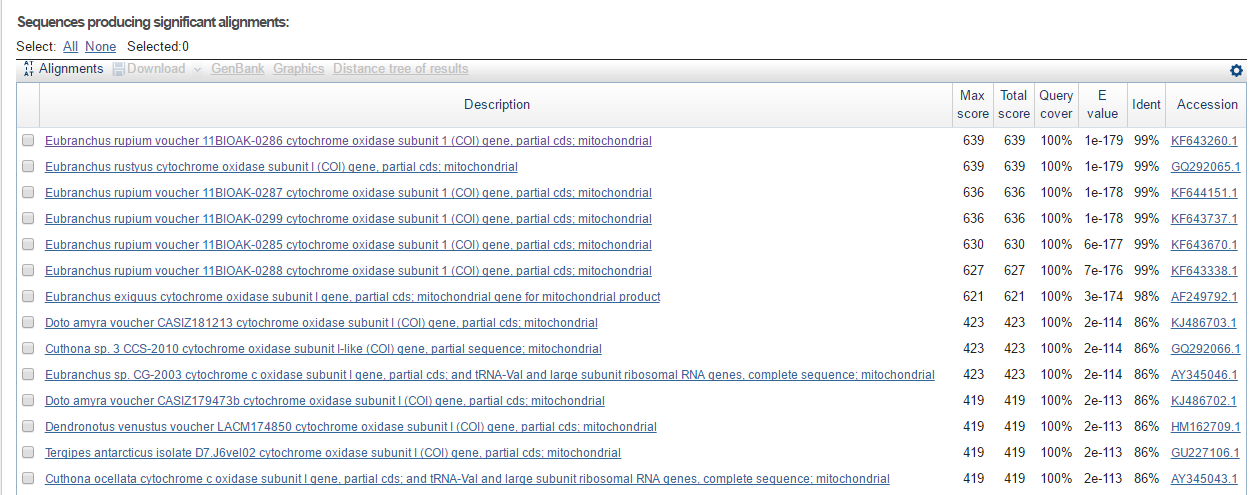 |
| Лучшие результаты blastn |
|---|
Изучаемая нами последовательность, вероятнее всего, является MT-CO1 геном – митохондриальным геном, кодирующим белок субъединицы 1 цитохром С оксидазы .
 Цитохром с-оксидаза (комплекс IV ) — терминальная оксидаза аэробной дыхательной цепи переноса
электронов, которая катализирует перенос электронов с цитохрома с на кислород с образованием воды.
Цитохромоксидаза присутствует во внутренней мембране митохондрий всех эукариот, а также в
клеточной мембране многих аэробных бактерий.
Цитохром с-оксидаза (комплекс IV ) — терминальная оксидаза аэробной дыхательной цепи переноса
электронов, которая катализирует перенос электронов с цитохрома с на кислород с образованием воды.
Цитохромоксидаза присутствует во внутренней мембране митохондрий всех эукариот, а также в
клеточной мембране многих аэробных бактерий.Комплекс IV из митохондрий млекопитающих и птиц состоит из 13 белковых субъединиц, три из которых обладают каталитической активностью, связывают кофакторы и кодируются генами митохондрий. Остальные десять субъединиц закодированы в ДНК ядра.
Субъединица 1, белок которой закодирован в нашей последовательности, является одной из трех больших субъединиц комплекса (I—III), которые несут на себе все необходимые кофакторы и осуществляют основные реакции катализа, связанные, в том числе, с переносом протонов. Как уже было упомянуто, она кодируется митохондриальными генами. К ее специфическим функциям относится связывание гема а, гема а3, центра CuB [1].
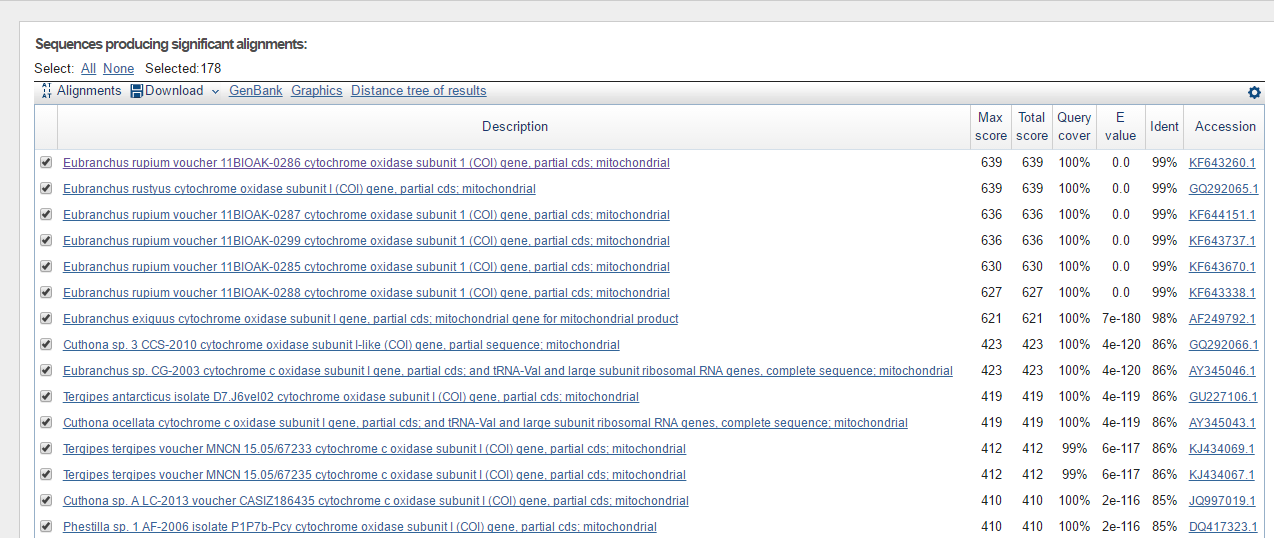
Из результатов работы blastn можно предположить таксономию прочтенной последовательности. Для этого я взяла первые 7 находок, так как они имели наилучшие и в целом очень хорошие значения Е-value, Query cover, Ident и Score, а для последущих находок вес был уже значительно ниже.
Источниками выбранных последовательностей являются гастроподы рода Eubranchus. При этом 5 из них относятся к виду Eubranchus rupium, 1 - Eubranchus rustyus и 1 - Eubranchus exiguus.
Для определения уровня таксономии было построено выравнивание из скачанных первых семи aligned sequences (alignedsequnces.txt). Видно, что последовательности практически идентичны, на данном участке есть всего несколько замен в пределах рода.

Ссылка на JalView проект
Таксономия до рода: Eukaryota; Metazoa; Lophotrochozoa; Mollusca; Gastropoda; Heterobranchia; Euthyneura; Nudipleura; Nudibranchia; Aeolididina; Aeolidioidea; Eubranchidae; Eubranchus.
Сравнение списков находок разными алгоритмами BLAST
Было призведено сравнение списков находок нуклеотидной последовательности тремя алгоритмами BLAST:
blastn, megablast и discontiguous megablast.
Чтобы результат сравнения был показателен, необходимо
было ограничить область поиска. Поиски как по роду, так и по семейству давали, на мой взгляд, слишком
мало находок (10 для megablast, 14 для discontiguous megablast и 18 для blastn). Когда область была
расширена до надсемейства (Aeolidioidea (taxid:71481)), а максимальное число находок
(Max target sequences) увеличено до 1000, находок blastn и discontiguous megablast оказалось слишком
много (618 и 621 соответсвенно). Чтобы получить удобное для сравнения число (несколько десятков)
мне пришлось провести поиск по надсемейсву Aeolidioidea (taxid:71481), исключив несколько семейств
(Aeolidiidae (taxid:195871), Favorinidae (taxid:252564), Glaucidae (taxid:216354))
и Uncultured/environmental sample sequences.
| Параметры запуска BLAST | |||||||
|---|---|---|---|---|---|---|---|
| Database | Max Target Sequences | Expect Threshold | Word Size | Max matches | Match/Mismatch Scores | Gap Costs | |
| megablast | Nucleotide collection (nr/nt) | 100 | 10 | 28 | 0 | 1, -2 | Linear |
| discontiguous megablast | Nucleotide collection (nr/nt) | 1000 | 10 | 11 | 0 | 2, -3 | Existence:5, Extention: 2 |
| blastn | Nucleotide collection (nr/nt) | 1000 | 10 | 11 | 0 | 2, -3 | Existence:5, Extention: 2 |
| Результаты выдачи megablast |
|---|
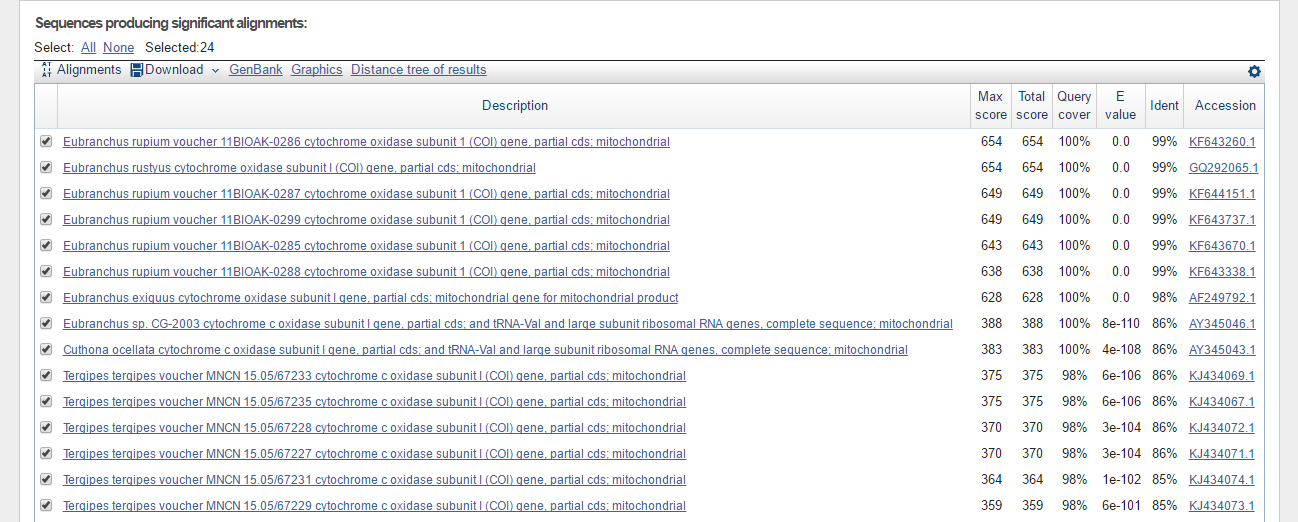 |
| Результаты выдачи discontiguous megablast |
|---|
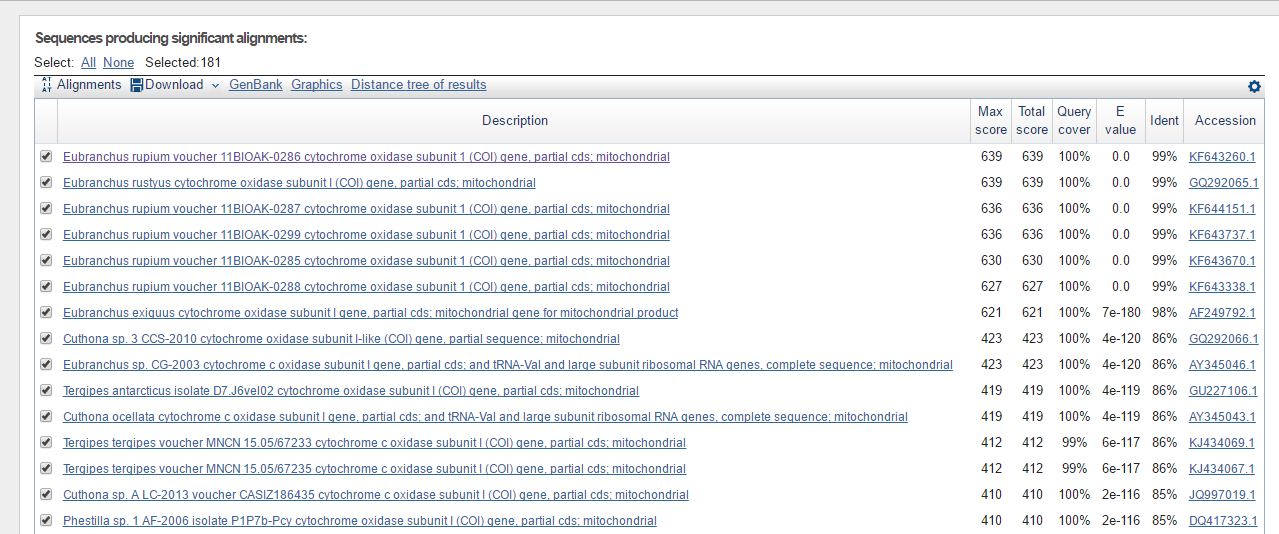 |
| Результаты выдачи blastn |
|---|
 |
| Сравнение алгоритмов | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Алгоритм | Число находок | Score лучшей находки | Score худшей находки | E-value лучшей находки | E-value худшей находки | Ident лучшей находки | Ident худшей находки | Query cover лучшей находки | Query cover худшей находки |
| megablast | 24 | 654 | 255 | 0.0 | 9e-70 | 99% | 80% | 100% | 100% |
| discontiguous megablast | 181 | 639 | 73.4 | 0.0 | 7e-15 | 99% | 67% | 100% | 72% |
| blastn | 178 | 639 | 134 | 0.0 | 3e-33 | 99% | 70% | 100% | 89% |
Больше всего находок выдал алгоритм discontiguous megablast.
Пример находки, найденной им и не найденной другими:
Pteraeolidia ianthina isolate Singapore B cytochrome oxidase subunit I (COI) gene, partial cds; mitochondrial (Max Score 73.4, Query Cover 72%, E-value 7e-15, Ident 67%).
Меньше всего находок дает алгоритм megablast.
Пример находки, найденной discontiguous megablast и blastn, но не найденной megablast:
Nanuca sebastiani cytochrome c oxidase subunit I (COI) gene, partial cds; mitochondrial (Max Score 374, Query Cover 97%, E-value 2e-105, Ident 84%).
Проведенное сравнение позволяет сделать вывод, что алгоритмы blastn и discontiguous megablast при использованных параметрах работают примерно одинаково и выдают не сильно отичающееся число находок сходного качества. Лучшие находки всех трех алгоритмов одинаковы.
Выбор алгоритма в каждом конкретном случае должен соответсвовать преследуемой цели.
Blastn предназначен для поиска не обязательно родственных, но похожих последовательностей. Среди найденных последовательностей могут быть и негомологичные, что необходимо учитывать при дальнейшем анализе. Поиск blastn сравнительно медленный, размер слова от 7 до 15.
(Cлово, инициирующее выравнивание - слово определенной длины, после нахождения которого blast начинает строить полное выранивание. Blast ищет совпадения слов не менее заданной длины между входной последовательностью и последователбностями из банка, и в случае нахождения такого слова начинает строить полное выравнивание последовательностей. Некоторые алгоритмы могут допускать mismatch.)
Discontiguous megablast подходит для межвидового поиска гомологов. Минимальный размер слова - 11, но допускается mismath.
Megablast работает гораздо строже, отсеивая большее количество находок и, следовательно, выдавая последовательности, лишь наиболее близкие к исходной. Он подходит для поиска близкородственных последовательностей, работает достаточно быстро. Размер слова не может быть ниже 16 нуклеотидов.
Проверка наличия гомологов белков
В этом задании было необходимо проверить наличие гомологов определенных белков в геноме организма X5 (Amoboaphelidium protococarum)
c помощью локального BLAST.
Для начала я создала локальную базу данных (makeblastdb -in X5.fasta -dbtype nucl), а затем
для каждого из выбранных белков запустила по ней алгоритм tblastn, находящий гомологи белка в формальной трансляции
нуклеотидного банка (tblastn -query xxx.fasta -db X5.fasta > xxx.out).
Белок HSP7C_HUMAN
HSP7C_HUMAN - консервативный шаперон HSP70, белок теплового шока. Имеется у большинства организмов из всех царств.
Играет роль репрессора активации транскрипции. Является компонентом PRP19-CDC5L комплекса, формирующего интегральную часть
сплайсосомы, и необходим для активации сплайсинга пре-мРНК. Связывая бактериальные ЛПС (липополисахариды), является посредником в процессах
ЛПС-индуцированного воспаления, включая секрецию моноцитами TNF (фактора некроза опухоли) [2].
Результаты tblastn по базе данных X5.fasta для данного белка: HSP7C_HUMAN.out.
| Всего находок - 16 |
|---|
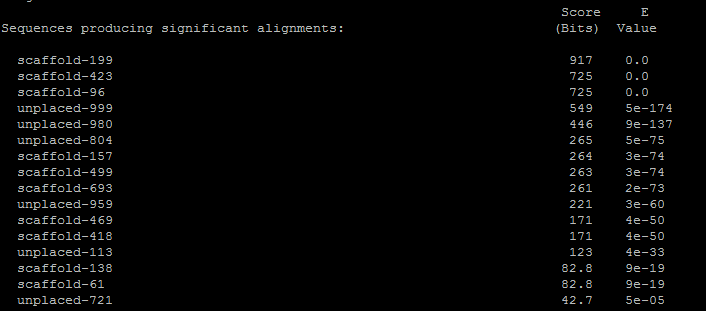 |
Лучшая находка имеет следующие параметры:
> scaffold-199
Length=1112851
Score = 917 bits (2369), Expect = 0.0, Identities = 474/607 (78%), Positives = 538/607 (89%), Gaps = 0/607 (0%), Frame = -2
У находки хороший E-value, достаточно высокие проценты Positives и Identities. На мой взгляд, параметры сходства достаточны, чтобы назвать ее гомологом исследуемого белка, вероятно выполняющим ту же функцию.
Белок TERT_HUMAN
TERT_HUMAN - теломераза, восстанавливающая длину хромосомы при репликации. Имеется у большинства,
но не у всех эукариот. Активна в прогениторных и раковых клетках, в нормальных же соматических не
активна или проявляет очень низкую активность. Играет важную роль в процессах старения и предотвращении
апоптоза. [3].
Результаты tblastn по базе данных X5.fasta для данного белка: TERT_HUMAN.out.
| Всего находок - 3 |
|---|
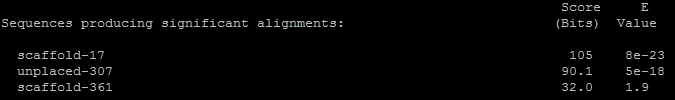 |
Лучшая находка имеет следующие параметры:
> scaffold-17
Length=2125590
Score = 105 bits (263), Expect = 8e-23, Identities = 151/568 (27%), Positives = 248/568 (44%), Gaps = 43/568 (8%), Frame = +1
Этот результат является условно положительным. BLAST выдал три находки, однако даже у самой лучшей из них параметры сходства слишком низкие, чтобы утверждать сохранение функций. Гомология отдельных доменов также маловероятна, так как совпадения распределены по всей длине последовательности относительно равномерно.
Белок CISY_HUMAN
CISY_HUMAN - митохондриальная цитратсинтаза. Участвует в цикле трикарбоновых кислот.
Проявляет каталитическую активность в реакции Acetyl-CoA + H2O + oxaloacetate = citrate + CoA.
[4].
Результаты tblastn по базе данных X5.fasta для данного белка: CISY_HUMAN.out.
| Всего находок - 4 |
|---|
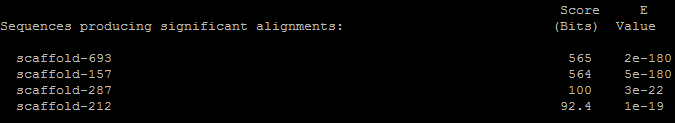 |
Лучшая находка имеет следующие параметры:
> scaffold-693
Length=1268102
Score = 565 bits (1457), Expect = 2e-180, Identities = 262/377 (69%), Positives = 315/377 (84%), Gaps = 3/377 (1%) Frame = +1
Скорее всего она действительно является гомологом интересующего нас белка с сохранением функций ввиду относительно хороших параметров сходства.
Поиск гена белка, закодированного в одном контиге ''Amoboaphelidium''
Для поиска я выбрала контиг unplaced-982. Его длина составляет 23575 п.н., следовательно, на нем
вполне может поместиться ген.
Информация о длинах контигов была получен командой infoseq
пакета EMBOSS: infoseq X5.fasta -only -name -length. Командой seqret X5.fasta: unplaced-982 -out
unplaced982.fasta я извлекла последовательность выбранного контига в отдельный файл
(unplaced982.fasta).
Далее был запущен blastn с параметрами по умолчанию и ограничению по таксону Amoeboaphelidium (taxid:1243176).
Результаты работы blastn:
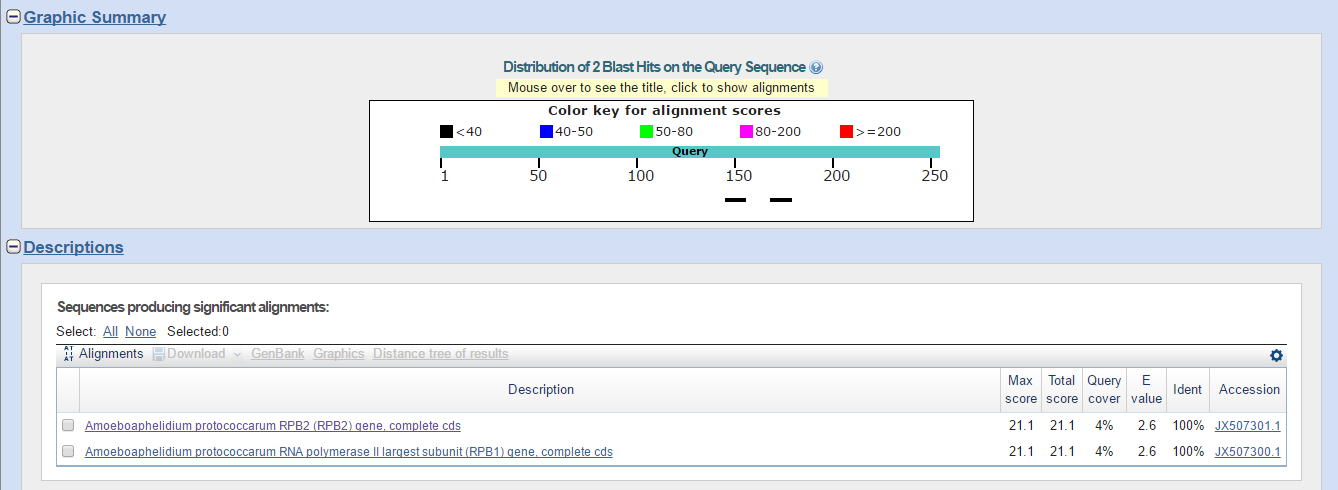 Как видно из рисунка, blastn выдает всего две находки, одинаковые по всем параметрам. Ident находок - 100%, однако
покрытие составляет всего 4% и E-value очень высок. Тем не менее, за неимением других,
мы все же можем предположить, что в контиге закодированы гены, указанные в данных находках.
Как видно из рисунка, blastn выдает всего две находки, одинаковые по всем параметрам. Ident находок - 100%, однако
покрытие составляет всего 4% и E-value очень высок. Тем не менее, за неимением других,
мы все же можем предположить, что в контиге закодированы гены, указанные в данных находках.
Оба гена кодируют белки болших субъединиц РНК-полимеразы II. Находка
Amoeboaphelidium protococcarum RNA polymerase II largest subunit (RPB1) gene, complete cds
- наибольшей субъединицы.
Находка
Amoeboaphelidium protococcarum RPB2 (RPB2) gene, complete cds - второй по величине субъединицы.