Построение дерева по нуклеотидным последовательностям
В этом задании было необходимо построить филогенетическое дерево выбранных ранее бактерий,
используя последовательности РНК малой субъединицы рибосомы (16S rRNA). Данные нуклеотидные
последовательности я получила из базы полных геномов NCBI. Для каждой из бактерий был открыт полный геном,
в нем были найдены координаты генов РНК 16S рибосомной субъединицы, которые затем были открыты в формате
fasta и собраны в единый fasta-файл (nuc_sequences.fasta).
Стоит отметить, что в геномах бактерий встречается несколько генов, кодирующих 16S-субъединицу. В каждом случае
последовательность для выравнивания была выбрана случайно.
С помощью сервера Muscle было получено выравнивание
последоваетельностей (nuc_ali.fasta).
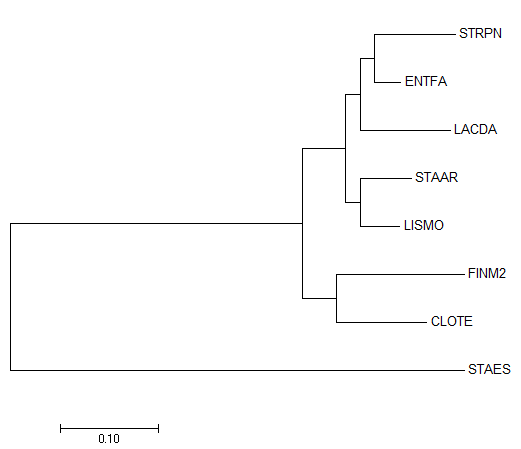
Выравнивание было открыто в мега методом Analyze, и по нему было реконструировано филогенетическое дерево с
использованием способа Maximum likelihood (Рис. 1.)
 |
 |
| Рис. 1. Дерево нуклеотидных последовательностей, построенное методом Maximum likelihood | Рис. 2. Эталонное дерево |
Полученное дерево содержит 5 нетривиальных ветвей:
- {ENTFA, STRPN} против {STAAR, STAES, LISMO, CLOTE, FINM2, LACDA}
- {ENTFA, STRPN, LACDA} против {STAAR, STAES, LISMO, CLOTE, FINM2}
- {STAAR, LISMO} против {ENTFA, STRPN, STAES, LACDA, CLOTE, FINM2}
- {CLOTE, FINM2} против {STAAR, STAES, LISMO, ENTFM, STRPN, LACDA}
- {CLOTE, FINM2, STAES} против {STAAR, LISMO, ENTFM, STRPN, LACDA}
Построение и анализ дерева, содержащего паралоги
В выбранных бактериях были найдены гомологи белка
CLPX_BACSU. Для этого сначала был создан
единый файл с протеомами бактерий proteomes.fasta.
По нему с помощью команды
makeblastdb -in proteomes.fasta -out db.fasta -dbtype prot
была создана база данных для
blastp.
Затем командой
blastp -query CLPX_BACSU.fasta -db db.fasta -evalue 0.001 -out
result.txt
был получен файл
results.txt, содержащий среди прочего мнемоники последовательностей, дающих с нашим белком
выравнивания с e-value не хуже заданного (0.001). Всего их оказалось 39.
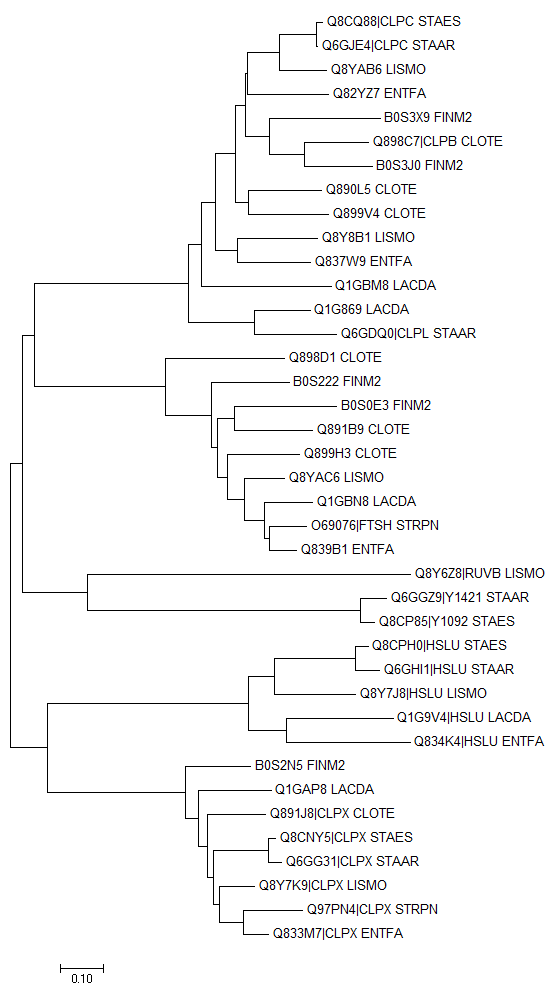 |
| Рис. 3. Дерево последовательностей гомологов, построенное методом Neighbour-joining |
Выравнивание было открыто в Mega, и по нему методом Neighbour-joining было построено филогенетическое дерево. (Рис. 3) Считая, что данное дерево реконструировано верно, можно попытаться найти на нем ортологов и паралогов.
Гомологичные последовательности называют ортологами, если они произошли в результате видообразования. Паралоги - гомологи, которые произошли в результате дупликации.
В нашем случае два гомологичных белка будем называть ортологами, если они:
а) из разных организмов;
б) разделение их общего предка на линии, ведущей к ним, произошло в результате видообразования.
Два гомологичных белка из одного организма будем называть паралогами.
На данном дереве примерами ортологов являются:
- Q8CQ88|CLPC STAES и Q6GJE4|CLPC STAAR
- Q97PN4|CLPX STRPN и Q833M7|CLPX ENTFA
- Q8Y8B1 LISMO и Q837W9 ENTFA
- Q1G9V4|HSLU LACDA и Q834K4|HSLU ENTFA
- Q890L5 CLOTE и Q899V4 CLOTE
- B0S3X9 FINM2 и B0S3J0 FINM2
- Q8YAB6 LISMO и Q8Y8B1 LISMO
- Q1G869 LACDA и Q1GBM8 LACDA
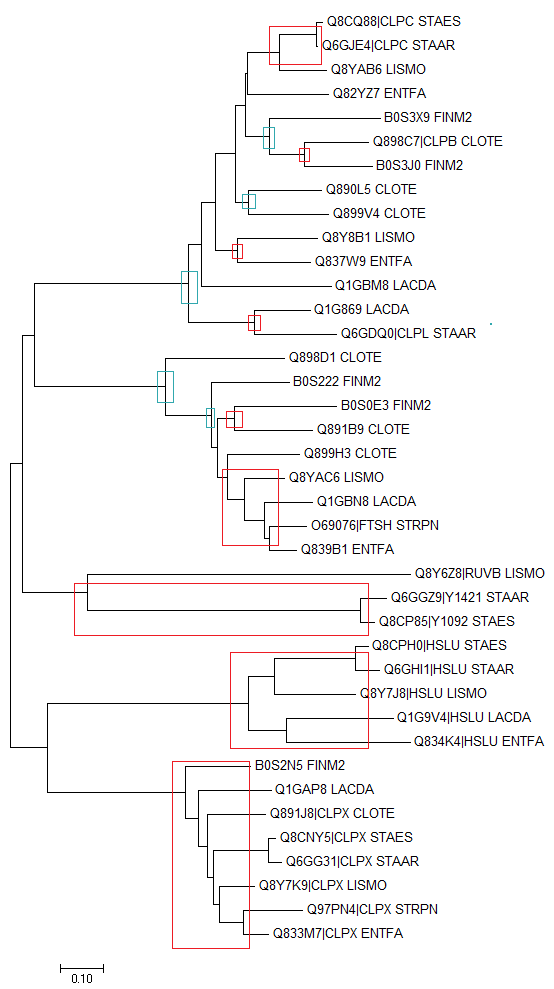 |
| Рис. 4. Примеры эволюционных событий |
Также по имеющемуся дереву можно попытаться восстановить эволюционные события. На рисунке 4 красными рамочками выделены некоторые примеры разделения путей эволюции в результате видообразования, а голубыми рамочками - дупликации генов.