Определение сайтов связывания транскрипционного фактора в участке хромосомы человека
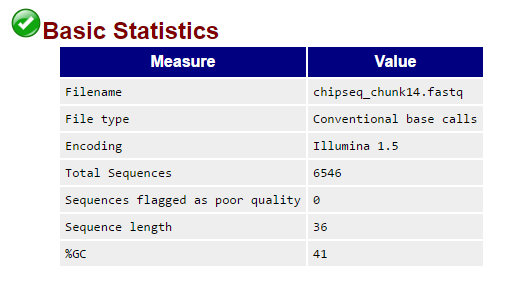 |
| Рис. 1. Информация о ридах в файле chipseq_chunk14.fastq |
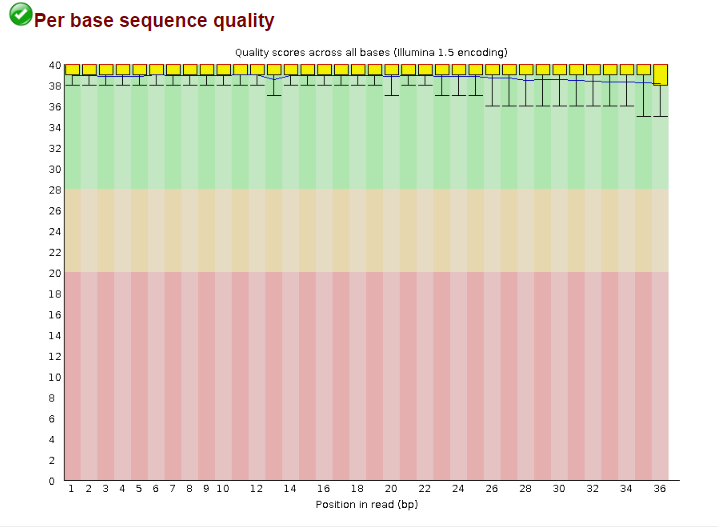 |
| Рис. 2. Per Base Sequence Quality, chipseq_chunk14.fastq |
Для начала с помощью программы FastQC был проведен контроль качества прочтений. Программа FastQC позволяет проводить быстрый и несложный контроль качества "сырых" данных секвенирования. Она проводит набор стандартных статистических анализов, которые можно использовать для предварительной оценки релевантности данных и выявления возможных проблем в полученной последовательности.
Использованная команда: fastqc chipseq_chunk14.fastq.
В результате работы программы был получен архив (.zip), содержащий отчет в виде файла fastqc_report.html.
Ссылка на страницу с отчетом: fastqc_report.html.
Всего в файле оказалось 6546 ридов, и ни один из них не был отмечен как некачественный (Рис. 1).
В целом на всем протяжении последовательности качество чтений очень хорошее, ни один из ящиков (включая усы) даже к концу последовательности не выходит за пределы зеленой области (области высокого качества) (Рис. 2). Поэтому в чистке с помощью Trimmomatic необходимости нет.
Далее последовательности были откартированы на проиндескированный геном человека.
Использованная команда:
bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk14.fastq > chipseq_chunk14.sam
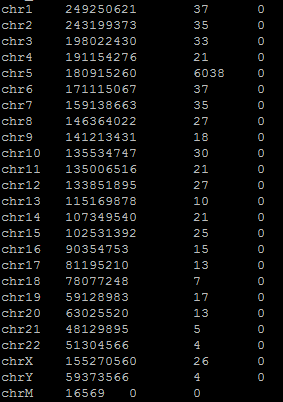 |
| Рис. 3. Откартированные риды |
Перевод в бинарный формат: samtools view -b chipseq_chunk14.sam -o chunk14.bam
Сортировка по координате в референсе начала чтения: samtools sort chunk14.bam chunk14_sorted.bam
Индексация: samtools index chunk14_sorted.bam
Получение информации и количестве откартированных ридов: samtools idxstats chunk14_sorted.bam
Всего в файле было 6546 ридов.
Все они откартировались на геном (Рис. 3). Подавляющее большинство ридов откартировались на chr5. Это позволяет предположить, что для анализа были предложены прочтения с пятой хромосомы.
Для поиска пиков я воспользовалась программой MACS. Она была запущена командой:
macs2 callpeak -n chunk14 -t chunk14_sorted.bam --nomodel
На выходе были получены следующие файлы: chunk14_peaks.xls, chunk14_summits.bed, chunk14_peaks.narrowPeak.
Все они содержат информацию о найденных пиках, наиболее полная информация представлена в файле *_peaks.xls.
Всего программа нашла 6 пиков. Все они находятся в одном регионе 5 хромосомы. Ширина пиков варьируется от 200 до 284 нуклеотидов.
| Имя | Начало | Конец | Ширина | abs_summit | pileup | -log10pvalue | -log10qvalue |
|---|---|---|---|---|---|---|---|
| chunk14_peak_1 | 77779687 | 77779970 | 284 | 77779779 | 26.00 | 20.42412 | 13.17144 |
| chunk14_peak_2 | 77826235 | 77826517 | 283 | 77826379 | 42.00 | 30.77239 | 21.30753 |
| chunk14_peak_3 | 77974151 | 77974405 | 255 | 77974279 | 22.00 | 15.04807 | 8.43140 |
| chunk14_peak_4 | 77995753 | 77995991 | 239 | 77995875 | 28.00 | 21.02363 | 13.74910 |
| chunk14_peak_5 | 78067225 | 78067471 | 247 | 78067333 | 21.00 | 18.16418 | 11.24452 |
| chunk14_peak_6 | 78439925 | 78440124 | 200 | 78440025 | 22.00 | 18.26694 | 11.30587 |
В данной таблице указаны показатели -log10pvalue и -log10qvalue. Чем выше данные параметры, тем ниже соответствующие показатели p-value и q-value, а следовательно, достовернее пик. Самым достоверным в нашем случае является пик 2, а наименее достоверным - пик 5.
Пики были визуализированы с помощью UCSC Genome Browser.
Использовался файл chunk14_peaks.narrowPeak, в начало которого предварительно была помещена следующая информация: track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="chunk14_peaks"
browser position chr5:77779600-78440400
Изображение пиков представлено на рисунке 4.
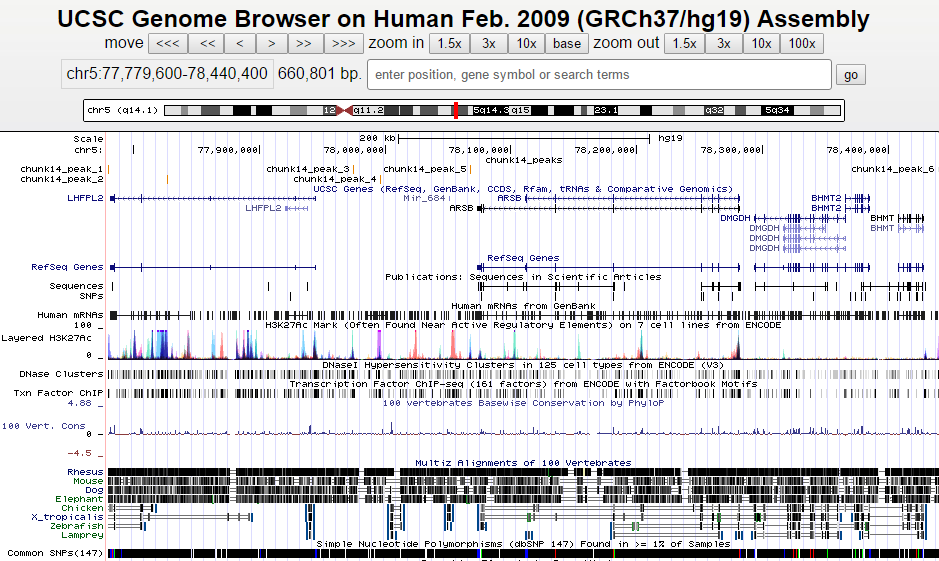 |
| Рис. 4. Визуализация пиков в UCSC Genome Browser |
Рассмотрим несколько пиков более подробно. Во-первых, хотелось бы рассмотреть наиболее достоверный пик, то есть пик 2, а также наиболее широкий пик, то есть пик 1.
| Имя | Начало | Конец | Ширина | -log10pvalue | -log10qvalue | Расстояние от старта до вершины |
|---|---|---|---|---|---|---|
| chunk14_peak_1 | 77779687 | 77779970 | 284 | 20.42412 | 13.17144 | 92 |
| chunk14_peak_2 | 77826235 | 77826517 | 283 | 30.77239 | 21.30753 | 144 |
Оба этих пика имеют достаточно хорошие показатели достоверности. Они являются и самыми достоверными, и самыми широкими среди остальных. На рисунках 5 и 6 представлены увеличенные изображения данных пиков в геномном браузере UCSC.
 |
| Рис. 5. Визуализация пика chunk14_peak_1 |
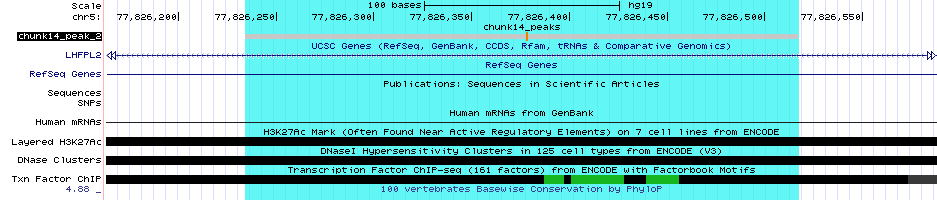 |
| Рис. 6. Визуализация пика chunk14_peak_2 |
Оба пика пересекают ген LHFPL2, имеющий координаты chr5:77,781,038-77,944,648. Он кодирует трансмембранный белок lipoma HMGIC fusion partner-like 2 protein, мутации в котором приводят к глухоте. При этом, как видно из рисунков, пик 2 попадает внутрь экзона, а пик 1 нет.
С помощью команды ../tools/seqtk/seqtk subseq ../hg19/GRCh37.p13.genome.fa chunk14_peaks.narrowPeak > chunk14_peaks.fasta файл chunk14_peaks.narrowPeak был переведен в формат fasta.
Полученнный файл: chunk14_peaks.fasta
Затем на основании полученного файла я попыталась построить выравнивание (Рис. 7).
 |
| Рис. 7. Выравнивание последовательностей пиков, окраска Clustalx |
Консенсусную последовательность (~ 4 - 8 nts) в нем идентифицировать не удалось.