Реконструкция эволюции доменной архитектуры
Выравнивание представителей домена Pfam белков с разной доменной архитектурой
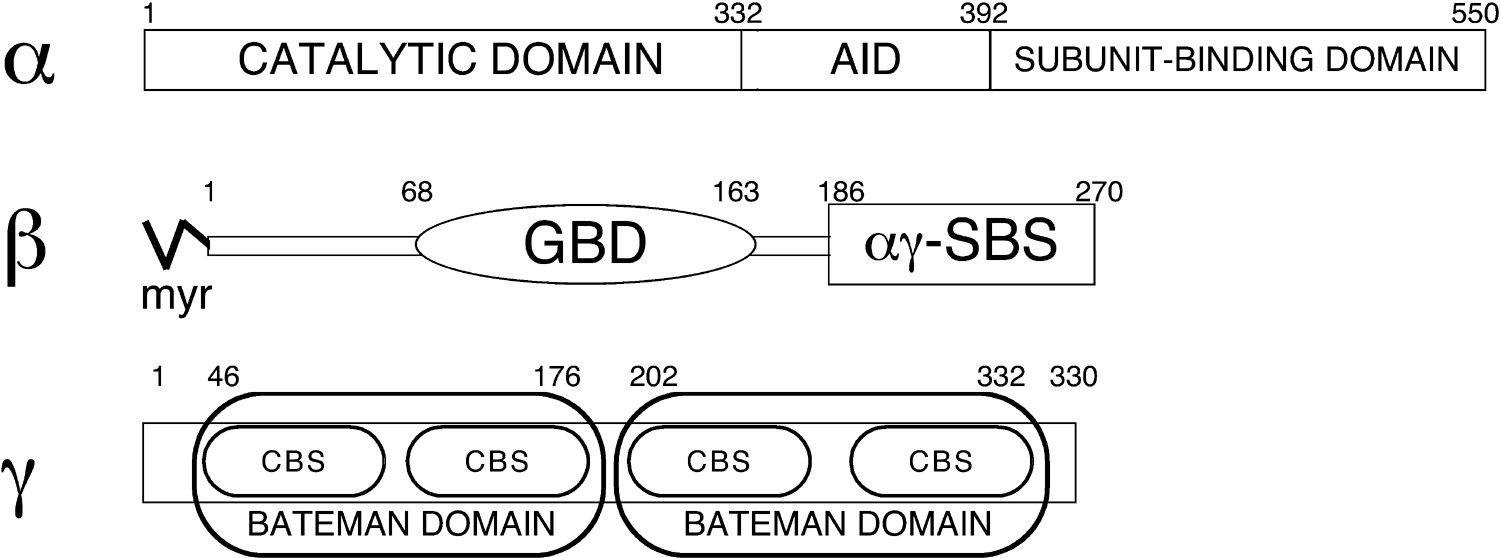
Для работы был выбран домен AMPK1_CBM
(ID: AMPK1_CBM; AC: PF16561) - сайт узнавания гликогена АМФ-активриуемой протеинкиназы
(АМФК).
АМФ-активируемая протеин-киназа - гетеротримерный комплекс, состоящий из каталитической
субъединицы α и 2 регуляторных субъединиц (β и γ), существующих во множестве изоформ и
сплайсинговых вариантов и в совокупности дающих 12 возможных гетеротримерных комбинаций.
АМФК активируется во множестве различных клеток в ответ на повышение концентрации АМФ и
обычно рассматривается как "метаболит-чувствительная киназа". Активация АМФК наблюдается
после теплового шока, усиленных физических упражнений, при гипоксии/ишемии, голодании.
При этом она фосфорилирует ключевые белки-мишени, контролирующие такие метаболические
процессы, как кетогенез в клетках печени, синтез холестерола, триглицеридов, липолиз
адипоцитов, окисление жирных кислот в мышцах и синтез белков. Хотя полноценная картина
регуляции активности АМФК еще не полностью ясна, практически уверенно можно утверждать,
что АМФК определяет чувствительность всего организма к инсулину и может предотвращать
появление инсулиновой резистентности частично путем ингибирования каскадов,
противодействующих инсулиновому сигналингу. [1]
 |
| Рис. 1. Домен AMPK1_CBM (GBD) в составе АМФК [2] |
В семействе Pfam данного домена 2886 последовательностей, они принадлежат 1058 видам. Домен входит в 124 различных архитектуры и для 56 последовательностей известна 3D структура.
Список разных доменных архитектур с доменом AMPK1_CBM можно найти по ссылке.
Для дальнейшей работы мною были выбраны архитектуры, представленные наибольшим числом последовательностей (Таблица 1).
| № | Доменный состав | Число представителей | Изображение |
| 1 | только домен AMPK1_CBM | 1204 |  |
| 2 | домен AMPK1_CBM и AMPKBI - связывающий домен β субъединицы АМФК | 1034 |  |
| 3 | домен AMPK1_CBM и DSPc - каталитический домен фосфатазы двойной специфичности | 153 |  |
| Таблица 1. Описание отдельных доменных архитектур, включающих домен AMPK1_CBM (зеленый) | |||
С помощью Jalview было построено выравнивание доменных участков всех белков, содержащих домен AMPK1_CBM (окраска BLOSUM62). Также была добавлена 3D структура одного домена. Для этого последовательность AAKB2_HUMAN была связана с PDB кодом 2F15.
Узнать, есть ли в моем случае 3D структура одного домена, а также увидеть соответствие между Uniprot ID и PDB кодами можно было в разделе Structures.
Ссылка на Jalview-проект:domain_ali.jvp.
Чтобы получить информацию об архитектуре всех последовательностей, содержащих домен AMPK1_CBM, я воспользовалась скриптом swisspfam-to-xls.py, который преобразует информацию о доменах в последовательности из файла swisspfam в таблицу для Excel. Файл swisspfam для всех последовательностей Uniprot скачан на kodomo. Чтобы получить информацию конкретно для моего домена, в качестве параметра -p был указан необходимый Pfam AC.
Использованная команда: python swisspfam_to_xls.py -z /srv/databases/pfam/swisspfam.gz -p PF16561 -o PF16561.xls
Полученный файл:PF16561.xls.
В Excel файл была добавлена сводная таблица, содержащая данные о том, какая последоввательность какой архитектуре принадлежит. Также в список последовательностей были добавлены колонки с информацией о таксономической принадлежности. Для этого из Uniprot были скачаны полные записи всех последовательностей в текстовом формате, а затем был запущен скрипт uniprot_to_taxonomy.py, принимающий на вход файл с Uniprot последовательностями и выдающий таблицу Excel с информацией о таксономии из полученных записей.
Использованная команда: python uniprot_to_taxonomy.py -i everything.txt -o taxonomy.xls
Полученная таксономия была перенесена в основную таблицу с помощью ВПР (VLOOKUP). Также туда была добавлена колонка с длиной выбранного домена из каждой последовательности.
Для последующей работы я выбрала таксон Eukaryota и два его подтаксона - Fungi и Metazoa. Последовательности из данных подтаксонов с выбранными архитектурами представлены на отдельном листе Excel файла (требовалось выбрать не менее 10 представителей архитектуры в каждом подтаксоне). Для архитектур 1 и 2 в выбранных подтаксонах оказалось более чем достаточно представителей, а вот архитектура 3 оказалась представлена практически исключительно в подтаксоне Viridiplantae. Поэтому далее работа проводилась только с архитектурами 1 и 2.
Итоговый Excel файл: final.xls.
Далее было построено выравнивание последовательностей только выбранных представителей архитектур 1 и 2. Для этого был использован скрипт filter_alignment.py, принимающий на вход fasta-файл с последовательностями и список имен тех последоваетльностей, которые необходимо извлечь. Для отделения имени последовательности от координат в домене в выравнивании Pfam была исользована опция -a "/".
Использованная команда: python filter-alignment.py -i all_ali.mfa -m ids -o my_ali.fasta -a "/"
Имена последовательностей в полученном файле были отредактированы таким образом, чтобы в них были закодированы доменные архитектуры и сравниваемые таксоны. Перед каждым ID было вставлено одно из следующих обозначений:
- M1 - подтаксон Metazoa, архитектура 1
- M2 - подтаксон Metazoa, архитектура 1
- F1 - подтаксон Fungi, архитектура 1
- F2 - подтаксон Fungi, архитектура 2
Затем "профильтрованное" выравнивание с измененными именами было открыто в Jalview и отредактировано. Были удалены пустые колонки и созданы 2 группы, соответсвующие архитетурам 1 и 2. В каждой группе отдельно была задана окраска по ClustalX. Несколько слишком коротких последовательностей и последовательностей, выровненных явно неправильно, были удалены. Исходя из последовательности с известной вторичной структурой (AAKB2_HUMAN) была добавлена разметка по вторичной структуре и температурному фактору.
Ссылка на итоговый Jalview проект: my_ali.jvp.
На мой взгяд, в полученном выравнивании нет каких-либо очевидных ошибок, однако в целом оно не такое хорошее, как я ожидала получить для консервативного домена. Тем не менее, вертикальные блоки удовлетворительного качества присутствуют, поэтому попробовать отреконструировать по этому выравниванию филогению все же (наверное) можно.
Построение филогенетического дерева домена
На основе выравнивания, содержащего последовательности общего для всех представителей домена,
в программе MEGA методом Neighbour Joining было построено филогенетическое дерево (Рис. 2).
Для подтверждения достоверности ветвей применялся Bootstrap тест со 100 репликами.
Изображение дерева было отредактировано с помощью сервиса ITOL.
Клады, включающие только представителей Metazoa, были окрашены красным, представителей Fungi -
зеленым. Листья с представителями архитектуры 1 покрашены черным, а архитектуры 2 - фиолетовым.
 |
| Рис.2 Дерево, постороенное методом Neighbour Joining c Bootstrap поддержкой ветвей |
На первый взгляд кажется, что получилось дерево, клады которого соответсвуют таксонам. В таком случае домен AMPK1_CBM, скорее всего, наследовался вертикально, у общего предка была только одна архитектура, предположительно 1, так как она более простая (хотя и не обязательно, ведь могла произойти и редукция), а архитектура 2 возникала несколько раз на разных ветвях.
Тем не менее, после того, как я укоренила дерево в среднюю точку программой retree, все оказалось не совсем так. Укоренение произошло не в ту ветвь, которая разделяет таксоны, хоть и достаточно близко к ней (Рис. 3).
 |
| Рис.3 Дерево, укорененное в среднюю точку |
Делать какие-либо предсказания на основании этого дерева уже гораздо сложнее, так как архитектуры домена на нем не образуют монофилетических групп достаточно высокого уровня. Однако на дереве есть некоторые "хорошие" клады, касательно которых можно что-то сказать. Так, например, верхняя окрашенная красным крупная клада, включающая только представителей Metazoa, четко разделяется на две более мелкие клады с разными доменными архитектурами. Это позволяет предполагать, что последний общий предок, с которого начинается данная крупная клада, уже имел обе доменные архитектуры. То же хотелось бы сказать и для нижней крупной клады, окрашенной зеленым и включающей только представителей Fungi, но здесь четкому разделению на две меньшие клады по доменным архитектурам "мешает" выбивающаяся последовательность A0A060SBD4_PYCCI. Возможно, это связано с ошибкой в построении дерева (что очень вероятно для деревьев доменов, ведь домены короткие, а мой - в особенности, так уж получилось). Также вероятно, что в данном месте действительно присутствует дополнительное разделение по архитектурам, а последовательность оказалась одна в силу ограниченности выборки. Однако в любом из этих случаев последний общий предок, находящийся в начале всей зеленой клады, вероятнее всего, тоже имел уже обе архитектуры, соответсвенно последующего усложнения/редукции не происходило.
Относительно остальных частей дерева, нам мой взгляд, нельзя утверждать ничего более менее конкретного, так как разделение по доменным архитектурам в них слишком дробное, и "клады" одинаковых доменных архитектур представлены всего 1-2 последовательностями. Соответсвенно и точная оценка числа перестроек доменной архитектуры на имеющемся дереве не представляется возможной.
В целом возникшие сложности в интерпритации результатов меня не удивили. Во-первых, выборка в нашем случае была явно недостаточно большой. Во-вторых, качество изначального выравнивания внушало сомнения. Наконец, ошибки в построении дерева тоже не исключены (хотя все три метода первой тройки выдали мне деревья с одинаковой топологией). Для более точной реконструкции филогении я бы попробовала увеличить выборку и применить другие спсобы укоренения, например с помощью аутгруппы.
Ссылки на скобочные формулы деревьев:
- Исходное дерево, построенное методом Neighbour Joining
- Дерево с Bootstrap поддержкой
- Дерево, укорененное в среднюю точку
Источники
[1]. Beate Fisslthaler, Ingrid Fleming, (2009) Activation and Signaling
by the AMP-Activated Protein Kinase in Endothelial Cells, Circulation Research
[2]. Polekhina G., Gupta A., van Denderen BJ, Feil SC, Kemp BE, Stapleton D,
Parker MW, (2005) Structural basis for glycogen recognition by AMP-activated protein kinase,
Elsevier