ПОДГОТОВКА ЧТЕНИЙ | ||||||||||||||||||||||||||||||||||||||||||
| В данном практикуме работа ведется с проектом по секвенированию бактерии Buchnera aphidicola (код доступа SRR4240389). Это короткие (длины 36) чтения, полученные по технологии Illumina. Файл был скачен из базы данных ENA, рапспакован с помощью команды: | ||||||||||||||||||||||||||||||||||||||||||
| gunzip SRR4240389.fastq.gz | ||||||||||||||||||||||||||||||||||||||||||
1. В первую очредь из полученного файла были удалены адапторы с помощью программы trimmomatic. Для этого использовалась следующая команда (где adapters.fasta - файл, содержащий последовательности адаптеров для Illumina): | ||||||||||||||||||||||||||||||||||||||||||
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 SRR4240389.fastq SRR4240389_clean.fastq ILLUMINACLIP:adapters.fasta:2:7:7 | ||||||||||||||||||||||||||||||||||||||||||
| После работы этой программы из 12950609 ридов, изначально содержавшихся в файле осталось 12947338 (99,97%), остальные 3271 (0,03%) были удалены, при этом по объему файл сократился на 1 Мбайт. | ||||||||||||||||||||||||||||||||||||||||||
| 2. После этого была проведена операция отрезания от ридов концевых нуклеотидов с Quality score меньше 20 (TRAILING:20) и длиной менее 30 (MINLEN:30) с помощью той же программы trimmomatic, команды: | ||||||||||||||||||||||||||||||||||||||||||
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 SRR4240389_clean.fastq SRR4240389_cleaner.fastq TRAILING:20 MINLEN:30 | ||||||||||||||||||||||||||||||||||||||||||
Как было показано ранее, такая операция существенно увеличивает качество прочтений. В результате работы программы из оставшихся 12947338 ридов было удалено более половины 6603310 (51,00%), осталось 6344028 (49,00%) прочтений. Размер файла сократился с 1386 Мбайт до 639 Мбайт. | ||||||||||||||||||||||||||||||||||||||||||
ПОДГОТОВКА K-МЕРОВ | ||||||||||||||||||||||||||||||||||||||||||
| Для подготовки k-меров (или слов длины k) длины 29 и 25 использовалась программа velveth. На выходе ее работы появилась новая папка с тремя файлами: log, roadmaps, sequences. | ||||||||||||||||||||||||||||||||||||||||||
| velveth velveth 29 -fastq -short SRR4240389_cleaner.fastq velveth velveth1 25 -fastq -short SRR4240389_cleaner.fastq | ||||||||||||||||||||||||||||||||||||||||||
CБОРКА НА ОСНОВЕ K-МЕРОВ | ||||||||||||||||||||||||||||||||||||||||||
| С помощтю программы velvetg из полученных файлов с k-мерами были осущетвлены построение ориентированного графа де Брайна и сборка генома: | ||||||||||||||||||||||||||||||||||||||||||
| velvetg velvethvelvetg velveth1 | ||||||||||||||||||||||||||||||||||||||||||
| Програма дает на выход файлы Graph, Pregraph, Lastgraph, contigs.fa (содержит последовательности всех контигов), stats.txt (содержит информацию о всех контигах, состоящих на вершинах графа). В Таблице 1 приведена основная информация об этих двух сборках: | ||||||||||||||||||||||||||||||||||||||||||
Таблица 1. Характеристика сборок.
| ||||||||||||||||||||||||||||||||||||||||||
Как видно из Таблицы 1. N50 на 400 больше у сборки с К-мером длины 29, что идет вразрез с длиной контига, большей для сборки с К=25, число вершин графа также больше для 25, ровно как и число ридов. Далее был произведен более подробный анализ контигов: в Таблице 2 приведены средние значения для контигов, посчитанные с помощью Excel и информация о самых длинных контигах. | ||||||||||||||||||||||||||||||||||||||||||
Таблица 2. Характеристика контигов (средние значения и самые длинные).
| ||||||||||||||||||||||||||||||||||||||||||
В Таблице 3 приведены данные об аномальном покрытии встречающимся в обеих сборках (превышение среднего значения в пять раз, или значение в пять раз меньше среднего), а также о контигах с аномально большим покрытием. Данные были взяты из файла stats.txt папки velveth, образованного после сборки k-меров. Как видно, существует множество контигов с аномально низким покрытием. Контиги с аномально высоким покрытием имеют длину 1, и даже не содержатся в файле contigs.fa, однако информация о них оказалась вместе с информацией о более крупных контигах в файле stats.txt. | ||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
АНАЛИЗ | ||||||||||||||||||||||||||||||||||||||||||
Далее с помощью программы megablast каждый из трёх самых длинных контигов из Таблицы 2 был сравнен с хромосомой Buchnera aphidicola (GenBank/EMBL AC — CP009253). В таблице 4 приведены соответствующие результаты. | ||||||||||||||||||||||||||||||||||||||||||
Таблица 4. Сравнение сборки k-29 c референсным геномом.
| ||||||||||||||||||||||||||||||||||||||||||
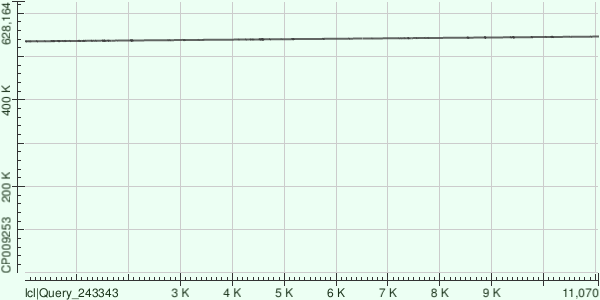
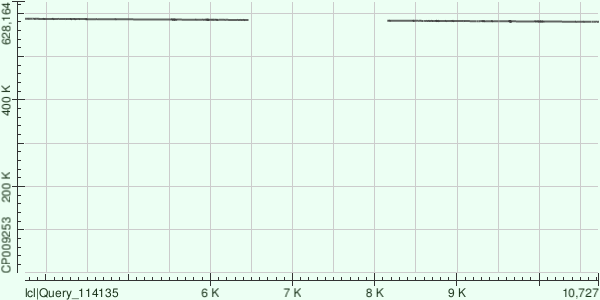
В первом случае было построено лишь одно выравнивание, для 62, 36 контига было построено 2 выравнивания, выбрано для таблицы было с большим max score. Ниже приведены иллюстрации полученных выравниваний, на которых видно, что в целом контиги ровно ложаться на хромосому, присутствую также небольшие делеции. | ||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||
СБОРКА ПОЛОВИНЫ РИДОВ | ||||||||||||||||||||||||||||||||||||||||||
С помощью программы tail была удалена первая половина очищенного от остатков адапторов, от концевых низкокачественных нуклеотидов и от коротких ридов файла SRR4240389_cleaner.fastq (были удалены 12 688 056 первых строчек): | ||||||||||||||||||||||||||||||||||||||||||
| tail -n 12 688 056 SRR4240389_cleaner.fastq>half_cleaner.fastq | ||||||||||||||||||||||||||||||||||||||||||
| Далее была произведена подготовка k-меров (k=2) и их сборка по алгоритму, описанному выше с помощью следующих команд: | ||||||||||||||||||||||||||||||||||||||||||
| velveth half_velveth 29 -fastq -short half_cleaner2.fastqvelvetg half_velveth/ | ||||||||||||||||||||||||||||||||||||||||||
| Ниже, в Таблице 5 приведены для сравнения характеристики двух проведенных сборок с k=29. Как видно, качетсво сборки существенно снизилось. | ||||||||||||||||||||||||||||||||||||||||||
Таблица 5. Сравнение характиристик сборки всех ридов и половины ридов.
| ||||||||||||||||||||||||||||||||||||||||||
Как видно из Таблицы 5. качество сборки сущестенно снизилось, параметр N50 уменьшился более чем в два раза, в два раза снизилась длина максимального контига, число контигов при этом фактичесски не изменилось, мало изменился и размер собранного генома. | ||||||||||||||||||||||||||||||||||||||||||
| Главнaя страница | ||||||||||||||||||||||||||||||||||||||||||