РЕСЕКВЕНИРОВАНИЕ. ПОИСК ПОЛИМОРФИЗМОВ У ЧЕЛОВЕКА.
I. ПОДГОТОВКА ЧТЕНИЙ |
В данном практикуме работа велась с чтениями экзома, картирующимися на участок хромосомы человека.
Файл chr16.fastq с одноконцевыми чтениями в формате fastq был взят из директории на kodomo /P/y14/term3/block4/SNP/reads .
Подготовка чтений осуществлялась в несколько этапов:
- Анализ качества прочтений
- Очистка чтений
- Сравнение параметров прочтений до и после очистки
|
АНАЛИЗ КАЧЕСТВА ПРОЧТЕНИЙ |
Анализ прочтений был осуществлен с помощью прогрммы FASTQC, установленной на сервере kodomo. Результатом работы программы является папка,
содержащая отчет о качестве прочтений, сопровожденный различными графическими изображениями, ознакомится с которыми можно ниже (Таблица 1., Рис.1, Рис.3, Рис.5, Рис.7, Рис.9).
Программа была запущена с помощью следующей команды: |
| fastqc chr16.fastq |
ОЧИСТКА ЧТЕНИЙ |
Очистка чтений проводилась с использованием программы Trimmomatic. Были удалены чтения с длиной
менее 50 нуклеотидов (параметр MINLEN:50) и с концов каждого чтения были отрезаны нуклеотиды с качеством ниже 20 (параметр TRAILING:20). Ниже
приведена команда: |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr16.fastq chr16_after.fastq TRAILING:20 MINLEN:50 |
СРАВНЕНИЕ ПАРАМЕТРОВ ЧТЕНИЙ ДО И ПОСЛЕ ЧИСТКИ |
| После очистки прочтений был проведен анализ полученного файла, с использованием команды описанной выше. Ниже, в Таблице 1 представлены основные
параметры прочтений до и после чистки, а на Рис. 1-10, полученных с помощью программы FASTQC, результаты представлены графически. |
Таблица 1. Характеристика прочтений до и после чистки.
| Параметр | Исходный файл | После чистки |
| Total Sequences | 3965 | 3798 |
| Sequences flagged as poor quality | 0 | 0 |
| Sequence length | 100 | 50-100 |
| %GC | 53 | 52 |
|
Как видно из Таблицы 1, количество прочтений после чистки уменьшилось на 167 ридов, видимо, за счет удаления ридов, длина которых
меньше 50 пн. |
Рис.1 График Quality scores для основания в чтении на данной позиции ДО ЧИСТКИ. 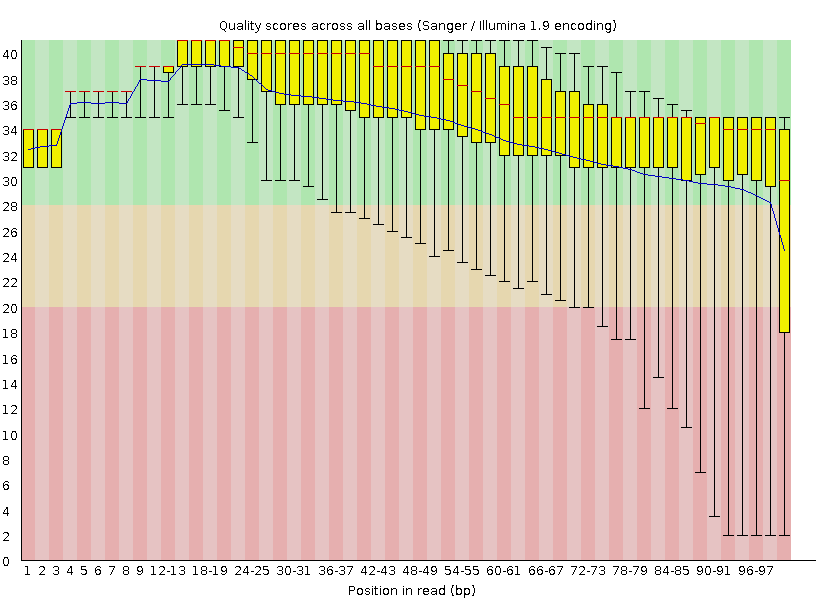
| Рис.2 График Quality scores для основания в чтении на данной позиции ПОСЛЕ ЧИСТКИ.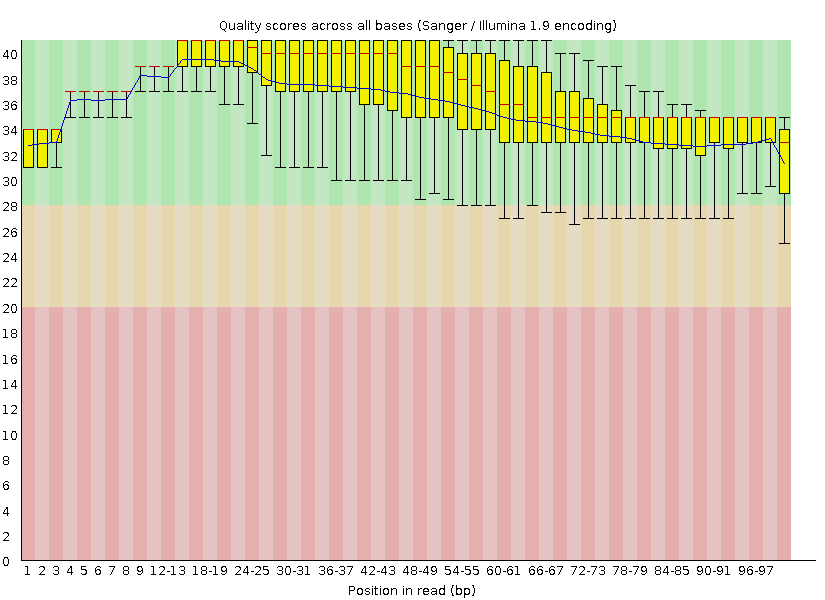 |
| Данные графики типа boxplot, показывают Quality scores (вертикальная ось) для каждой позиции прочтений (горизонтальная ось). При этом
красная линия отображает медиану разброса значений Quality scores среди оснований всех ридов находящихся на данной позиции, голубая линия - среднее, желтые столбцы
ограничены верхним и нижним квартилями, "усы" отражают значения 10ого и 90ого процентиля. Красная область показывает позиции, характеризующиеся низким
Quality score (<28), оранжевая средним, зеленая высоким (>20).
Как можно отметить, до очистки чтения обладают достаточно низкими параметрами: из 55 столбцов, у 19 90ый процентиль заходит в орнжевую зону,
у 14 в красную. По данному графику, что качество чтений низкое.
После чистки ситуация существенно поменялась: за счет отрезания с концов ридов нуклеотидов с Quality scores меньше 20, этот параметр существенно возрос,
в результате чего почти все столбцы полностью оказались в зеленой зоне, за исключением 17, "верхние усы" которых расположились в оранжевой зоне. |
Рис.3 График процентного содержания четерых типов нуклеотидов в данной позиции прочтений ДО ЧИСТКИ.
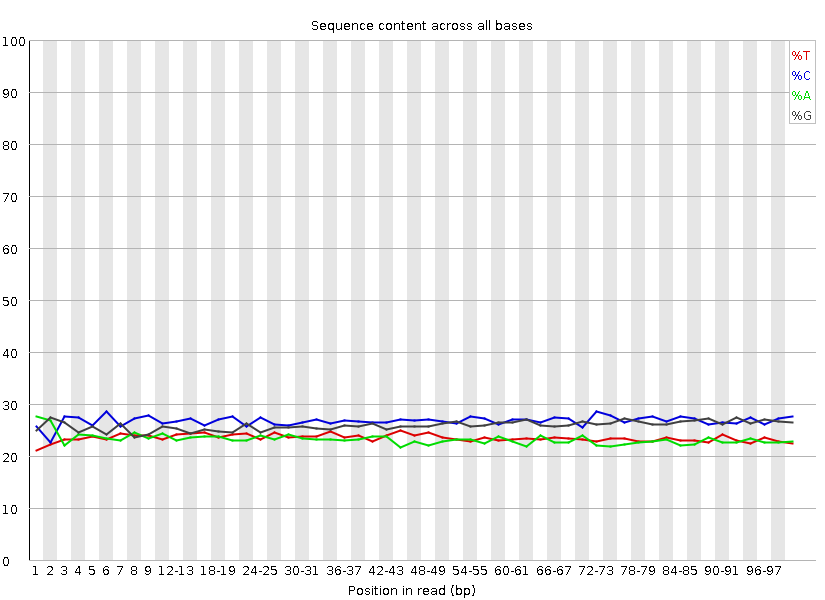 |
Рис.4 График процентного содержания четерых типов нуклеотидов в данной позиции прочтений ПОСЛЕ ЧИСТКИ.
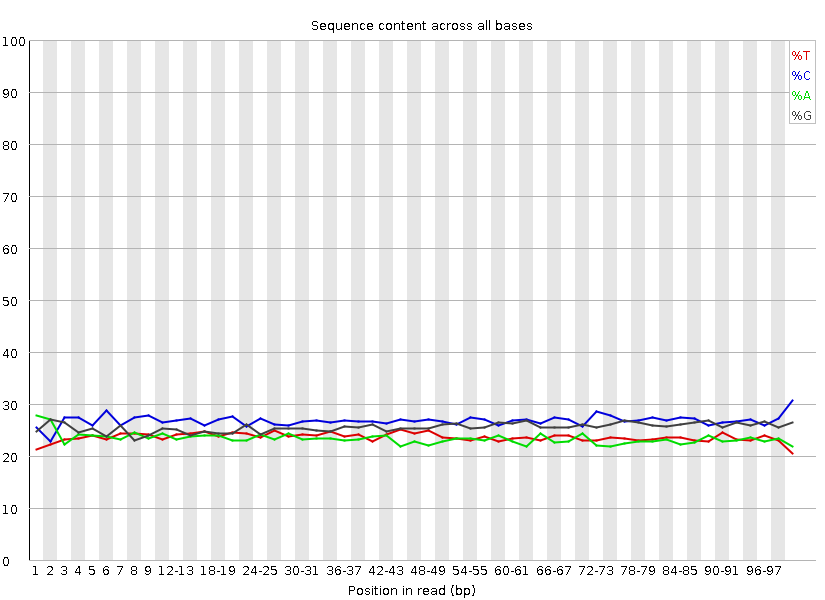 |
| Данные графики показывает какой процент занинает нуклеотид данного типа среди всех нуклеотидов встречающихся в данной позиции
ридов. На вертикальной оси отложен процент встречаемости, на горизонтальной - позиция.
Как видно, очистка не сильно повлияла на данный параметр, что согласуется с данными представленными в Таблице 1. Однако этот параметр
согласно руководству далек от идеального результата. |
Рис.5 График распределения соотношения GC% по всем прочтениях ДО ЧИСТКИ.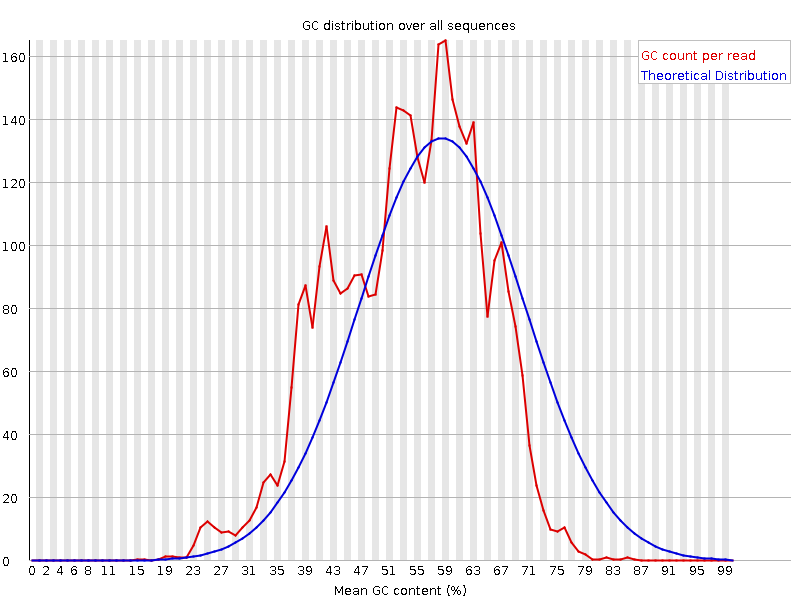 |
Рис.6 График распределения соотношения GC% по всем прочтениях ПОСЛЕ ЧИСТКИ.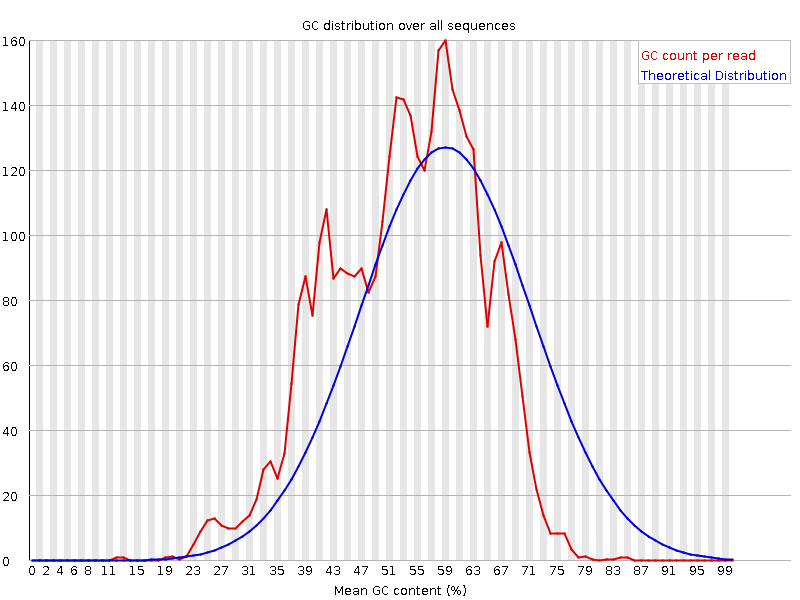 |
| Данные графики показывает насколько отличается распределение процентного содержания пары GC в риде от теоретически рассчитанного.
На горизонтальной оси отложен GC%, на вертикальной число ридов с таким значением GC%.
Чистка существенно не повлияла на распределение этого параметра. В обоих случаях практическое распределение довольно сильно отличается от теоретически
рассчитанного. |
Рис.7 График распределения Quality score всего прочтиния ДО ЧИСТКИ.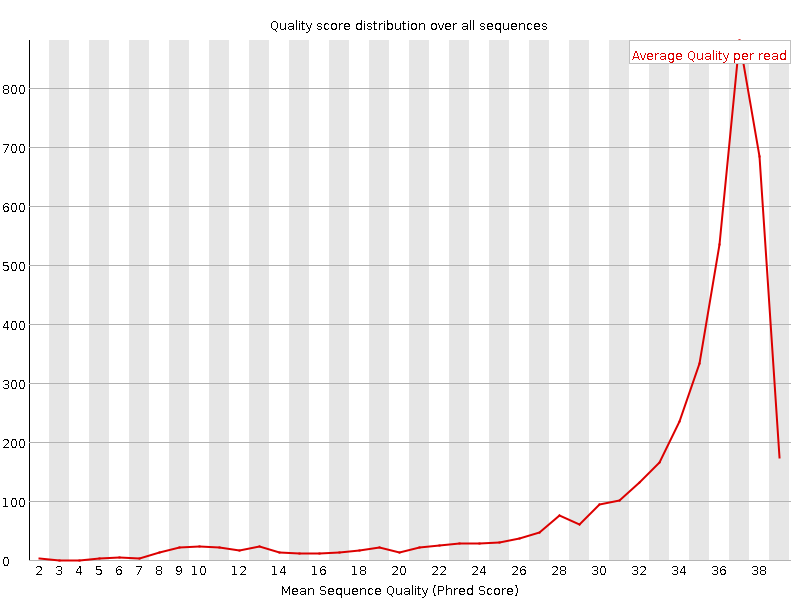 |
Рис.8 График распределения Quality score всего прочтиния ПОСЛЕ ЧИСТКИ.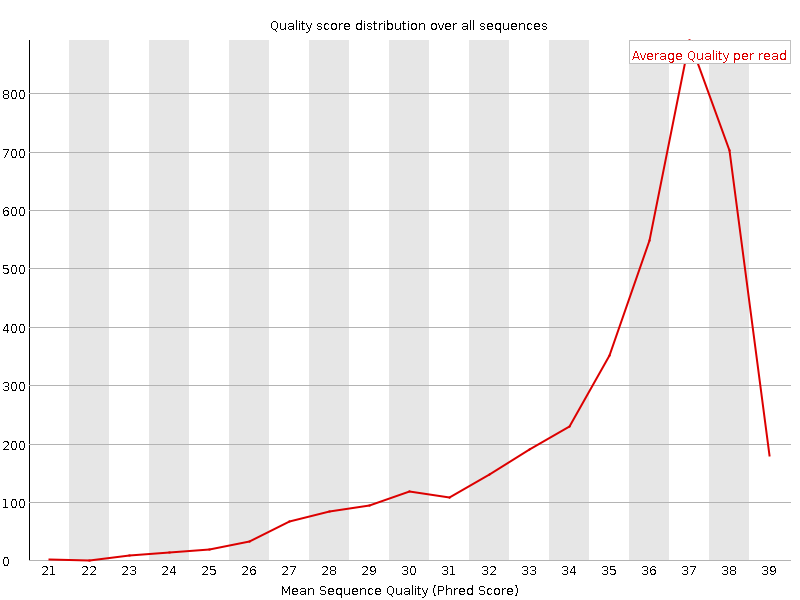 |
| Данные графики показывает распределение Quality score прочтений. По горизонтальной оси отложены значения Quality score прочтений,
по вертикальной число прочтений в котором такое значение встречается.
Как видно, после чистки, засчет отрезаня нуклеотидов с низким значением Quality score, график существенно поменялся. После чистки в графике
отсутствуют значения Quality score ниже 20. |
Рис. 9 График распределения длины прочтений ДО ЧИСТКИ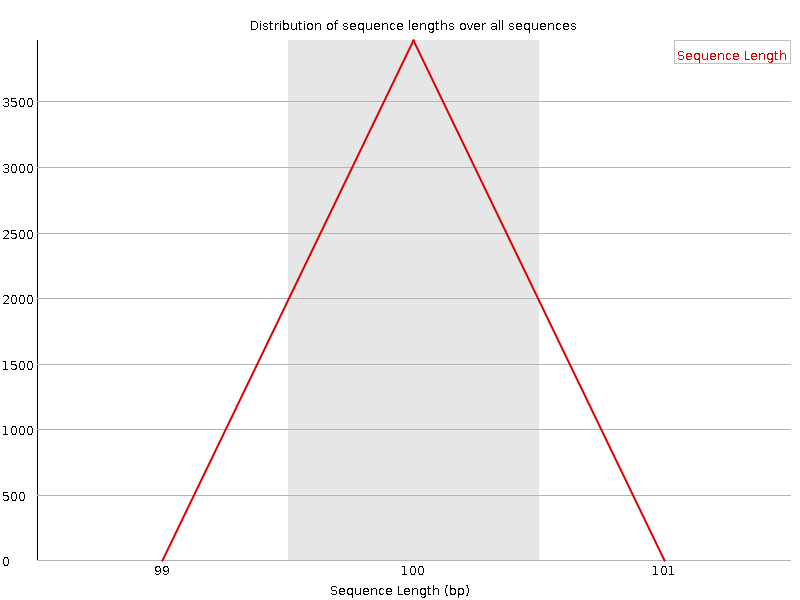 |
Рис. 10 График распределения длины прочтений ПОСЛЕ ЧИСТКИ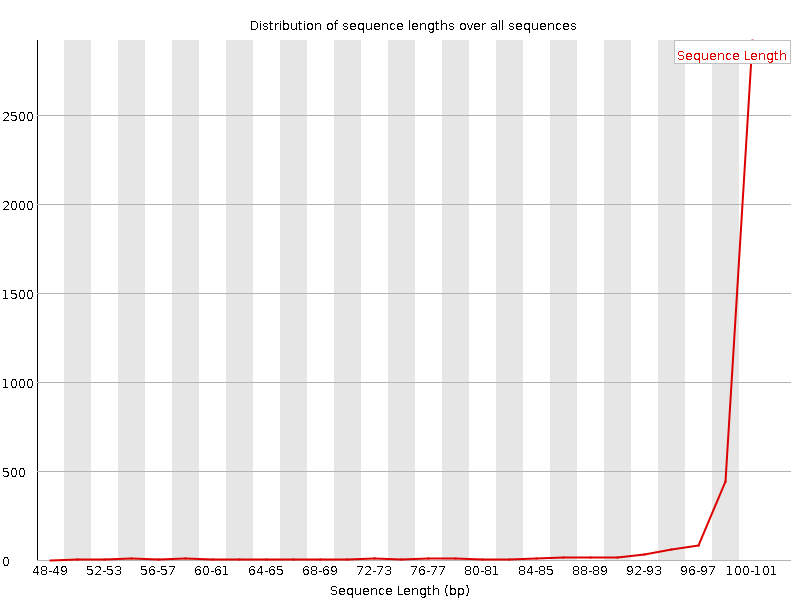 |
| На данном графике изображено распределение длин прочтений. По горизонтальной оси отложены длины прочтений, по вретикольной - чило
прочтений с этими длинами.
После чистки график очень существенно поменялся. Если до чистки длина прочтений в среднем составляла 100 пн, после чистки за счет удаления концевых нуклеотидов
стала колебаться от 50 до 100. |
|
II.КАРТИРОВАНИЕ ЧТЕНИЙ |
КАРТИРОВАНИЕ ЧТЕНИЙ |
Очищенные чтения были откратрированы с помощью программы Hisat2 в несколько этапов:
1.Экспорт программы в текущую директорию |
| export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 |
| 2.Индексирование референсной последовательности
|
| hisat2-build chr16.fasta chr16 |
| 3.Построение выравнивания прочтений и референса в формате .sam |
| hisat2 -x chr16 -U chr16_after.fastq --no-spliced-alignment --no-softclip > alignment1_chr16.sam |
АНАЛИЗ ВЫРАВНИВАНИЯ |
| 1. Полученный файл с выравниванием был переведен в бинарный формат .bam c использованием программы из пакета samtools - samtools view: |
| samtools view alignment1_chr16.sam -bo alignment1_chr16.bam |
| 2. Выравнивание чтений с референсом было отсортировано по координате в референсе начала чтения при помощи программы samtools sort: |
| samtools sort alignment1_chr16.bam -T temporary.txt -o sorted1.bam |
| 3. Командой samtools index полученный файл был проиндексирован: |
| samtools index sorted1.bam |
Выход программы Hisat2 |
Согласно данным полученным на выходе из программы Hisat2, из 3798 неповторяющихся прочтений:
- 33 прочтения (0.87%) не участвовали в построении выравниваия
- 3763 прочтения (99.08%) были выравнены ровно один раз
- 2 (0.05%) выравнивались более одного раза.
- Значение overall aligment rate - 99.13%
|
Определение среднего значения покрытия экзона чтениями |
| 1. С помощью команды samtools depth было вычислено покрытие чтениями для каждого нуклеотида. С помощью функции МАКС из пакета Excel
было найдено максимальное значение покрытия - 208 для нуклеотида 11362870. |
| samtools depth sorted1.bam>sorted_depth.txt |
| 2. С использованием GenomeBrowser для 16 хромосомы версии Hg19, был обнаружен экзон в состав которого входит данный нклеотид и
его координаты. |
Рис.11 Ген и Экзоны в состав которых входит нуклеотид 11362870. |
| 3. Для каждого нуклеотида входящего в экзон вновь были посчитаны покрытия. С помощью функции СРЗНАЧ пакета Excel было посчитано среднне покрытие
для данного экзона, которое составило 139,735. |
| depth -r chr16:11362720-11363119 sorted1.bam>exon_depth.txt |
III. АНАЛИЗ SNP |
ПОИСК SNP И ИНДЕЛЕЙ |
| 1. С помощью программы samtools mpileup -uf был создан файл с полиморфизмами в формате .bcf: |
| samtools mpileup -uf chr16.fasta sorted1.bam >snp.bcf |
| 2. Файл со списком отличий между референсом и чтениями в формате .vcf был создан с использованием команды "bcftools call -cv" пакета bcftools: |
| bcftools call -vc snp.bcf >snp.vcf |
| 3. Полученный файл содержал информацию о 65 полиморфизмах, 1 из которых явлеяется инделем, остальны 64 - единичные замены. Ниже в Таблице 2. приведены
данные о трех полиморфизмах. |
Таблица 2. Описание трех полиморфизмов.
| Координата | Тип полиморфизма | Референс | Прочтение | Глубина прочтения | Качество чтения |
| 56969148 | Замена | G | A | 80 | 225.009 |
| 56968938 | Замена | С | A | 3 | 3.5427 |
| 11444454 | Делеция | gaaaaaaaaaaa | gaaaaaaaaaa,gaaaaaaaaa | 55 | 10.8299 |
|
| Покрытие найденных SNP варьируется в пределах от одного до 145, среднее значение достигает 17,5625. Качество ридов начинается от 3,5427
и заканчивается 225,009, в среднем составляет 76,87, что является очень высоким показателем. Можно ознакомиться с файлом snp.vcf, полученным на выходе программы,
содержащим все значения. |
АННОТАЦИЯ SNP |
С помощью программы annovar полученные полиморфизмы были проаннотированы по следующим базам данных: |
- Refgene
- Dbsnp
- 1000 genomes
- GWAS
- Clinvar
|
Первоначльно, для работы с программой annovar необходимо было перевести файл в формат, подходящий для работы программы: |
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp.vcf -outfile snp.avinput |
После, с помощью annovar snp были проаннотриованы по базам данных. Ниже представлены команды для получения аннотаций и результат их работы в
краткой форме. |
1. REFGENE |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -out rs.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/ |
На выходе работы программы были получены три файла (rs.refgene.exonic_variant_function - информация о полиморфизмах в экзонах;
rs.refgene.log - информация о процессе работы программы; rs.refgene.variant_function - информация о локализации полиморфизма по отношению к функциональным
частям генома), сождержащих информацию о полиморфизмах, которая в кратком виде представлена в Таблицах 3, 4, 5. |
Таблица 3. Локализация полиморфизмов в геноме
| Категория | Пояснение | Количество SNP |
| downstream | snp за пределами гена | 2 |
| upstream | snp за пределами гена | 5 |
| UTR5 | snp в 5' нетранслируемой области | 3 |
| UTR3 | snp в 3' нетранслируемой области | 3 |
| intronic | snp в интроне | 24 |
| exonic | snp в экзоне | 9 |
| intergenic | snp входящая в несколько перекрывающихся генов | 19 |
|
Как видно из Таблицы 3, из всех полиморфизмов лишь небольшое количество содержится в экзонах. |
Всего snp попали в 8 генов, ниже приведена таблица, указывающая сколько snp в них располагается. |
Таблица 4. Гены, содержащие snp.
| Ген | Число Snp | Область | Функция гена (Кодирует:) |
| SOCS1 | 1 | 1 downstream | Cупрессор сигнального цитокина 1 |
| TNP2 | 4 | 1 экзон, 3 интрона | Переходный ядерный белок сперматид 2 |
| PRM3 | 5 | 2 экзона, 3 upstream | Протамин 3 |
| PRM2 | 4 | 2 экзона, 1 UTR3, 1 downstream | Протамин 2 |
| PRM1 | 21 | 1 экзон, 19 интегральных, 1 upstream | Протамин 1 |
| RMI2 | 35 | 1 экзон, 19 интегральных, 1 UTR5, 2 UTR3, 12 интронов | RecQ-mediated genome instability protein 2 |
| PRSS53 | 9 | 1 экзон, 8 интронов | Серин-протеаза 59 |
| HERPUD1 | 5 | 1 экзон, 1 интрон, 1 upstream, 2 UTR5 | Homocysteine-responsive endoplasmic reticulum-resident ubiquitin-like domain member 1 protein |
|
Все вышеперечисленные гены - белок-кодирующие, их белки выполняют совершенно различные функции:
- SOCS1 вовлечен в неготивную регуляцию цитокинов, которые учавствуют в регуляции JAK/STAT3 пути. Через связывание JAKs, SOCS1 ингибирует их киназную активность.
- TNP2 играет ключевую роль в замене гистонов на протамин в удлиненных сперматидах млекопитающих. В конденсирующих сперматидах, загружаемых на нуклеосомы, где он способствует обработке протаминов, которые ответственны за выселение гистонов
- PRM1/2/3 - протамины замещают гистоны в хроматине сперматозойдов во время гаплоидной фазы сперматогенза. Они компактизируют ДНК сперматозойдов в высококонденсированный, стабильный и неактивный комплекс.
- RMI2 - необходимый компонент RMI комплекса,который играет важную роль в процессинге интермедиатов гомологичной рекомбинации. Необходим для регулирования сегрегации сестренских хроматид и ограничения ДНК кроссовера.
- PRSS53 - in vitro может разоушать альфа-цепи фибриногена так же хорошо как плазминоген активатор про-урокиназного типа.
- HERPUD1 - компонент система контроля качества эндоплазматического ретикулума (ERQC), вовлечен в убхитин- зависимую деградацию неправильно уложенных белков эндоплазматического ретикулума.
|
В Таблице 5 предствалена информация о snp, содержащихся в экзонах, о качестве произошедшей в низ замены, об аминокислотной замене. |
Таблица 5. Характериситка snp в экзонах.
| Ген | Координата | Референс | Прочтение | Качество | Покрытие | Синонимичность замены | Аминокислотная замена |
| TNP2 | 11362729 | G | A | 225.009 | 145 | Несинонимичная | R131W |
| PRM3 | 11367143 | G | A | 221.999 | 35 | Нонссенс-мутация (появление стоп-кодона) | R104X |
| PRM3 | 11367154 | C | T | 221.999 | 30 | Несинонимичная | R100Q |
| PRM2 | 11369938 | G | A | 125.008 | 27 | Несинонимичная | P97L |
| PRM2 | 11369947 | G | A | 139.008 | 27 | Несинонимичная | A94V |
| PRM1 | 11374866 | G | T | 221.999 | 38 | Синонимичная | R47R |
| RMI2 | 11444572 | A | C | 225.009 | 113 | Синонимичная | T123T |
| PRSS53 | 31096164 | G | C | 148.077 | 7 | Несинонимичная | P406A |
| HERPUD1 | 56969148 | G | A | 225.009 | 80 | Несинонимичная | R50H |
|
|
2.DBSNP |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.snp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/ |
На выходе программа дает три файла, один из которых (rs.snp.log) содержит информацию о работе программы, другой (rs.snp.hg19_snp138_dropped) содержит данный о референсних SNP,
характеризующихся пометкой rs, третий (rs.snp.hg19_snp138_filtered) с snp данной отметкой не обладающими.
Из 65 полиморфизмов 7 не помечены rs, для остальных 48 присваивается rs идентификатор, можно ознакомится с файлом rs.snp.hg19_snp138_dropped, содержащим информацию об rs идентификаторе,
заменах, их коордикатах, покрытии и качесстве. |
3.1000 GENOMES |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.1000genomes -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/ |
Аналгично двум предыдущим командам, данная дает на выдачу три файла:
- rs.1000genomes.log - информация о работе программы
- rs.1000genomes.hg19_ALL.sites.2014_10_dropped - информация о частоте встречаемости snp, содержащихся в базе данных
- rs.1000genomes.hg19_ALL.sites.2014_10_filtered - спиок snp, не содержащихся в базе данных
|
Из 65 полиморфизмов 7 не встречаются в базе данных 1000 genomes, 48 была присвоена частота встречаемости, с которой можно ознакомится
в файле rs.1000genomes.hg19_ALL.sites.2014_10_dropped, полученном в ходе роботе программы.
Наиболее высокая частота встречаемости среди данных полиморфизмов - 0,984026 (замена, расположенная в интроне гена PRSS53),
самая низкая - 0,00499201 (замена, расположенная в интроне гена RMI2).Среднее значение для частоты встречаемости - 0,464222799. В таблице 6 приведена информация о частоте полиморфизмов, встречающихся
экзонах. |
Таблица 6. Встречаемость snp, локализованных в экзонах.
| Ген | Встречаемость | Референс | Прочтение | Координата | Синонимичность |
| TNP2 | 0.333267 | G | А | 11362729 | Несинонимичная |
| PRM3 | 0.956669 | G | A | 11367143 | Нонссенс-мутация (появление стоп-кодона) |
| PRM3 | 0.770168 | C | T | 11367154 | Несинонимичная |
| PRM2 | 0.0289537 | G | A | 11369938 | Несинонимичная |
| PRM2 | 0.0289537 | G | A | 11369947 | Несинонимичная |
| PRM1 | 0.487819 | G | T | 11374866 | Синонимичная |
| RMI2 | 0.178115 | A | C | 11444572 | Синонимичная |
| PRSS53 | 0.479034 | G | C | 31096164 | Несинонимичная |
| HERPUD1 | 0.148762 | G | A | 56969148 | Несинонимичная |
Любопытно, что snp, приводящая к образованию стоп-кодона в гене обладает очень высокой частотой встречаемости (0.956669). В целом частота
встречаемости данных полиморфизмов в экзонах довольно высока. |
4.GWAS |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out rs.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/ |
На выходе программы было получено два файла. Один из них (rs.gwas.log) содержит информацию о работе программы, другой (rs.gwas.hg19_gwasCatalog)содержат информаию о snp,
вызывающих фенотипические проявления. Этот файл rs.gwas.hg19_gwasCatalog содержит информацию о трех из 65 snp,
вызывающих различные фенотипические проявления. В таблице 7 представлена информация об snp, вызывающих фенотипические проявления и их локализации в геноме. |
Таблица 7. Встречаемость snp, локализованных в экзонах.
| Ген | Проявление | Замена | Локализация | Синонимичность | Координата | Встречаемость | Качество |
| PRM1 | Черты, связанные с проявлением ожирения | G -> T | Экзон | Сининимичная | 11374866 | 0.487819 | 221.999 |
| PRSS53 | Болезнь Паркинсона | C -> T | Интрон | - | 31095171 | 0.479233 | 221.999 |
| HERPUD1 | Метаболический синдром | G -> A | Экзон | Несинонимичная | 56969148 | 0.148762 | 225.009 |
Как видно из таблицы, из 65 snp встречаются три, вызывающие довольно яркие фенотипические проявления:
- Метаболический синдром - увеличение массы висцерального жира, снижение чувствительности периферических тканей к инсулину и гиперинсулинемия, которые нарушают углеводный, липидный, пуриновый обмен, а также вызывают артериальную гипертензию.
*Ассоциирован с spn в гене HERPUD1, ответственным за производство белка, осуществляющего контроль качества ЭР и деградацию неправильно уложенных белков ЭР.
- Болезнь Паркинсона - медленно прогрессирующее хроническое неврологическое заболевание, характерное для лиц старшей возрастной группы. Относится к дегенеративным заболеваниям экстрапирамидной моторной системы.
*Ассоциирована с spn в гене PRSS53, кодирующем белок Серин-протеаза 59.
- Черты, присущие ожирению
*Ассоциированы с spn в гене PRM1, кодирующем протамин 1.
Довольно любопытно, что полиморфизм, ассоциированный с развитием болезни Паркинсона, согласно базе данных Refgene расположен внутри интрона.
Не менее любопытное замечание заключается в том, что черты, связанные с ожирением ассоциированы с синонимичной заменой (!).Также, из таблицы видно,
что полиморфизма, ассоциированные с проявлением данных фенотипов довольно распространены. |
5.CLINVAR |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.clinvar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/ |
Программа дает на выход три файла:
- rs.clinvar.log - содержат данные о работе программы
- rs.clinvar.hg19_clinvar_20150629_dropped- содержат данные об аннотированных в базе данных проявлениях snp
- rs.clinvar.hg19_clinvar_20150629_filtered
|
К сожалению, по результату работы программы оказалось, что ни одна из описынных ранее 65 snp не аннотирована в базе данных Clinvar, и
файл rs.clinvar.hg19_clinvar_20150629_dropped оказался пуст. |
РЕЗУЛЬТАТЫ |
Ниже, В Таблице 8 представленны все команды, использовавшиеся в данном практикуме. В Таблице 9 собраны данны по всем базам дынных для полученных
65 snp. |
Таблица 8. Использванные команда
| Команда | Действие |
| fastqc chr16.fastq | Анализ качества прочтений |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr16.fastq chr16_after.fastq TRAILING:20 MINLEN:50 | Очистка чтений (отрезание концевых нуклеотидов с качеством ниже 20 и удаление чтений длиной меньше 50) |
| hisat2-build chr16.fasta chr16 | Индексирование референсной последовательности |
| hisat2 -x chr16 -U chr16_after.fastq --no-spliced-alignment --no-softclip > alignment1_chr16.sam | Построение выравнивания прочтений и референса в формате .sam |
| samtools view alignment1_chr16.sam -bo alignment1_chr16.bam | Переведение файла в бинарный формат .bam |
| samtools sort alignment1_chr16.bam -T temporary.txt -o sorted1.bam | Сортирует выравнивание чтений с референсом по координате в референсе |
| samtools index sorted1.bam | Индексирует файл |
| samtools depth sorted1.bam>sorted_depth.txt | Вычисляет покрытие для каждого нуклеотида |
| depth -r chr16:11362720-11363119 sorted1.bam>exon_depth.txt | Вычисляет покрытия для нуклеотидов данного промежутка (экзона в этом случае) |
| samtools mpileup -uf chr16.fasta sorted1.bam >snp.bcf | Создает файл с полиморфизмами в формате .bcf |
| bcftools call -vc snp.bcf >snp.vcf | Создает список отличий между референсом и чтениями в формате .vcf |
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp.vcf -outfile snp.avinput | Переводит .vcf файл в формат .avinput |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -out rs.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/ | Пойск аннотаций заданных snp в базе данных Refgene |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.snp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/ | Пойск аннотаций заданных snp в базе данных Refgene dbsnp |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.1000genomes -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/ | Пойск аннотаций заданных snp в базе данных 1000 genomes |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out rs.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/ | Пойск аннотаций заданных snp в базе данных GWAS |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.clinvar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/ | Пойск аннотаций заданных snp в базе данных Clinvar |
|
Таблица 9. Аннотации snp по 5 базам данных. Красным, Желтым и зеленым помеченны snp, аннотированные в базе данных 1000 genoms,
связанные с появлением определенных фенотипов (последняя колонка, расшифровка под таблицей).
| Ген | rs | Координата | Зона | Замена | Qscore | Depth | Частота | Синоним. | Аминок. | Фен. |
| SOCS1 | rs8060459 | 11348273 | downstream | T -> A | 4,1 | 10 | 0.007987 | - | - | - |
| TNP2 | rs8043625 | 11361895 | intronic | C -> G | 225 | 50 | 0,340855 | - | | |
| TNP2 | rs181695 | 11362554 | intronic | T -> C | 103 | 30 | 0,977236 | - | | |
| TNP2 | rs56069754 | 11362640 | intronic | G -> A | 167 | 50 | 0,164137 | - | | |
| TNP2 | rs11640138 | 11362729 | exonic | G -> A | 225 | 145 | 0,333267 | nonsyn. | R131W | |
| PRM3 | rs438289 | 11367143 | exonic | G -> A | 222 | 35 | 0,956669 | stopgain | R104X | |
| PRM3 | rs429744 | 11367154 | exonic | C -> T | 222 | 30 | 0,770168 | nonsyn. | R100Q | |
| PRM3 | rs2301127 | 11367477 | upstream | G -> A | 222 | 14 | 0,593251 | - | | |
| PRM3 | rs2857764 | 11367907 | upstream | G -> C | 55 | 6 | 0,959265 | - | | |
| PRM3 | rs74007622 | 11368053 | upstream | G -> A | 7 | 1 | 0,006989 | - | | |
| PRM2 | rs4781055 | 11369428 | downstream | C -> T | 109,2 | 9 | 0,154553 | - | | |
| PRM2 | rs424908 | 11369534 | UTR3 | G -> A | 222 | 49 | 0,967053 | - | | |
| PRM2 | rs74007625 | 11369938 | exonic | G -> A | 125 | 27 | 0,028954 | nonsyn. | P97L | |
| PRM2 | rs74007626 | 11369947 | exonic | G -> A | 139 | 27 | 0,028954 | nonsyn. | A94V | |
| PRM1 | rs737008 | 11374866 | exonic | G -> T | 222 | 38 | 0,487819 | Synonim. | R47R | 1 |
| PRM1 | rs74007629 | 11375209 | upstream | G -> A | 87 | 9 | 0,007388 | - | | |
| PRM1,RMI2 | rs74731751 | 11376499 | intergenic | C -> T | 4,8 | 1 | 0,007588 | - | | |
| PRM1,RMI2 | rs55707838 | 11378075 | intergenic | C -> T | 11,3 | 1 | 0,155351 | - | | |
| PRM1,RMI2 | rs11074957 | 11378151 | intergenic | G -> A | 7,8 | 1 | 0,480232 | - | | |
| PRM1,RMI2 | - | 11391899 | intergenic | G -> T | 11,3 | 1 | - | - | | |
| PRM1,RMI2 | rs7206320 | 11393685 | intergenic | A -> G | 10,4 | 1 | 0,684904 | - | | |
| PRM1,RMI2 | rs55726383 | 11394552 | intergenic | C -> A | 10,4 | 1 | 0,011981 | - | | |
| PRM1,RMI2 | - | 11394652 | intergenic | G -> T | 7 | 1 | - | - | | |
| PRM1,RMI2 | rs77305343 | 11400991 | intergenic | A -> C | 8,6 | 1 | 0,011182 | - | | |
| PRM1,RMI2 | rs7190580 | 11403470 | intergenic | A -> G | 4,8 | 1 | 0,783946 | - | | |
| PRM1,RMI2 | rs1345878 | 11404753 | intergenic | A -> G | 6,2 | 1 | 0,769169 | - | | |
| PRM1,RMI2 | rs74007667 | 11409945 | intergenic | C -> T | 11,3 | 1 | 0,006789 | - | | |
| PRM1,RMI2 | rs9927945 | 11412772 | intergenic | T -> C | 7,8 | 1 | 0,969649 | - | | |
| PRM1,RMI2 | rs9302459 | 11420339 | intergenic | G -> T | 7,8 | 1 | 0,805112 | - | | |
| PRM1,RMI2 | - | 11420402 | intergenic | G -> T | 5,5 | 1 | - | - | | |
| PRM1,RMI2 | rs8049090 | 11421095 | intergenic | G -> A | 19,8 | 2 | 0,373403 | - | | |
| PRM1,RMI2 | rs4390613 | 11422836 | intergenic | T -> C | 9,5 | 1 | 0,807708 | - | | |
| PRM1,RMI2 | rs370573625 | 11429529 | intergenic | C -> T | 4,8 | 1 | 0,186102 | - | | |
| PRM1,RMI2 | rs9922790 | 11435422 | intergenic | T -> G | 10,4 | 1 | 0,96865 | - | | |
| PRM1,RMI2 | rs11645404 | 11436629 | intergenic | G -> A | 11,3 | 1 | 0,184505 | - | | |
| RMI2 | rs12149160 | 11439303 | UTR5 | G -> T | 32,8 | 2 | 0,360623 | - | | |
| RMI2 | rs918737 | 11439639 | intronic | T -> C | 15,9 | 2 | 0,510982 | - | | |
| RMI2 | rs74010208 | 11439665 | intronic | C -> G | 3,5 | 2 | 0,004992 | - | | |
| RMI2 | rs918738 | 11439679 | intronic | C -> G | 44,8 | 2 | 0,377596 | - | | |
| RMI2 | rs7184280 | 11441012 | intronic | A -> G | 7 | 1 | 0,96905 | - | | |
| RMI2 | rs13331623 | 11441498 | intronic | T -> C | 11,3 | 1 | 0,471645 | - | | |
| RMI2 | rs7189044 | 11442196 | intronic | G -> C | 3,5 | 2 | 0,213458 | - | | |
| RMI2 | rs7191093 | 11442279 | intronic | A -> G | 5,5 | 1 | 0,178115 | - | | |
| RMI2 | rs2032929 | 11444403 | intronic | T -> C | 177 | 27 | 0,886382 | - | | |
| RMI2 | rs28627609 | 11444425 | intronic | C -> T | 61 | 25 | 0,213259 | - | | |
| RMI2 | rs2032930 | 11444430 | intronic | T -> G | 222 | 30 | 0,887181 | - | | |
| RMI2 | - | 11444454 | intronic | * -> | 10,8 | 55 | - | - | | |
| RMI2 | rs2032931 | 11444469 | intronic | C -> T | 222 | 92 | 0,873602 | - | | |
| RMI2 | rs7204628 | 11444572 | exonic | A -> C | 225 | 113 | 0,178115 | synonim. | T123T | |
| RMI2 | rs6498185 | 11444726 | UTR3 | C -> T | 72 | 26 | 0,172324 | - | | |
| RMI2 | rs2032933 | 11445040 | UTR3 | G -> A | 11,3 | 1 | 0,885982 | - | | |
| PRSS53 | rs11865038 | 31095171 | intronic | C -> T | 222 | 53 | 0,479233 | - | | 2 |
| PRSS53 | rs11864806 | 31095204 | intronic | G -> C | 222 | 37 | 0,479034 | - | | |
| PRSS53 | rs11864839 | 31095251 | intronic | G -> C | 124 | 31 | 0,471446 | - | | |
| PRSS53 | rs7199949 | 31096164 | exonic | G -> C | 148,1 | 7 | 0,479034 | nonsyn. | P406A | |
| PRSS53 | - | 31097046 | intronic | G -> T | 11,3 | 1 | - | - | | |
| PRSS53 | rs4889535 | 31097574 | intronic | G -> C | 73 | 3 | 0,983027 | - | | |
| PRSS53 | rs74015064 | 31098223 | intronic | G -> A | 11,3 | 8 | 0,006989 | - | | |
| PRSS53 | rs6565218 | 31098582 | intronic | G -> T | 109,1 | 8 | 0,984026 | - | | |
| PRSS53 | rs73530203 | 31099859 | intronic | G -> A | 22 | 4 | 0,351438 | - | | |
| HERPUD1 | - | 56965947 | upstream | G -> T | 4,1 | 3 | - | - | | |
| HERPUD1 | rs37025 | 56966011 | UTR5 | G -> A | 53 | 5 | 0,758786 | - | | |
| HERPUD1 | rs37024 | 56966071 | UTR5 | T -> G | 49 | 4 | 0,609026 | - | | |
| HERPUD1 | - | 56968938 | intronic | C -> A | 3,5 | 3 | - | - | | |
| HERPUD1 | rs2217332 | 56969148 | exonic | G -> A | 225 | 80 | 0,148762 | nonsyn. | R50H | 3 |
|
| * - Индель: "gaaaaaaaaaaa - > gaaaaaaaaaa,gaaaaaaaaa"
1. - Фенотип: черты, присущие ожиреню
2. - Фенотип: Болезнь Паркинсона
3. - Фенотип: Метаболический синдром |
|
Главнaя страница
|
© Анна Камышева 2017
|