I. ПОДГОТОВКА ЧТЕНИЙ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| В данном практикуме работа велась с одноконцевыми чтениями, картирующимися на участок хромосомы человека, полученными в
результате секвенирования транскриптома (человек, версия сборки генома hg19).
Файлы chr16.1.fastq и chr16.2.fastq (две биологические реплики одноконцевых чтений в формате fastq) были взяты из директории на kodomo /P/y14/term3/block4/SNP/rnaseq_reads. Использовалась также разметка человеческого генома по версии Gencode19 для сборки hg19, взятая из директории /P/y14/term3/block4/SNP/rnaseq_reads Подготовка чтений осуществлялась в несколько этапов:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
АНАЛИЗ КАЧЕСТВА ПРОЧТЕНИЙ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
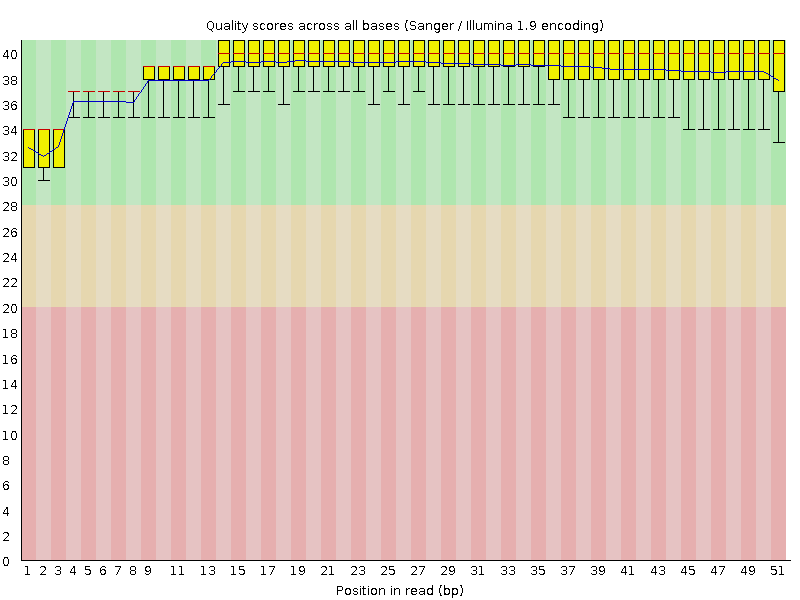
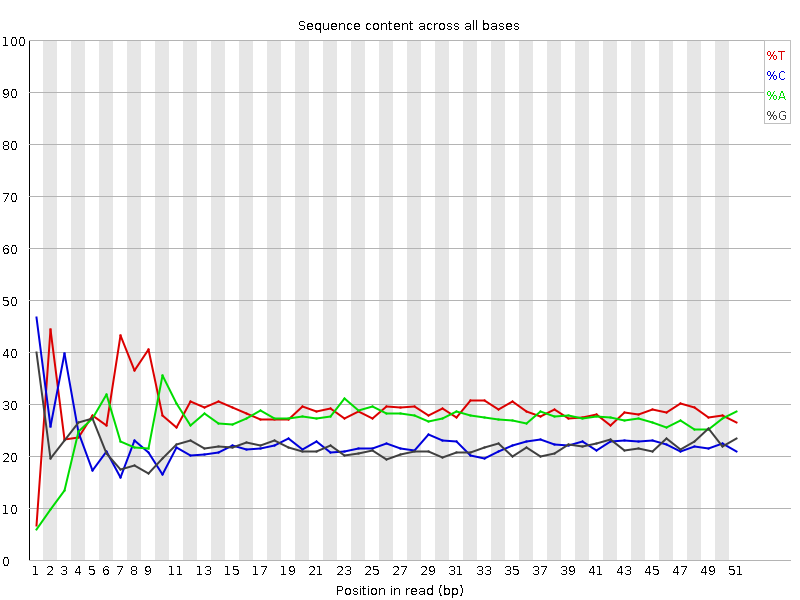
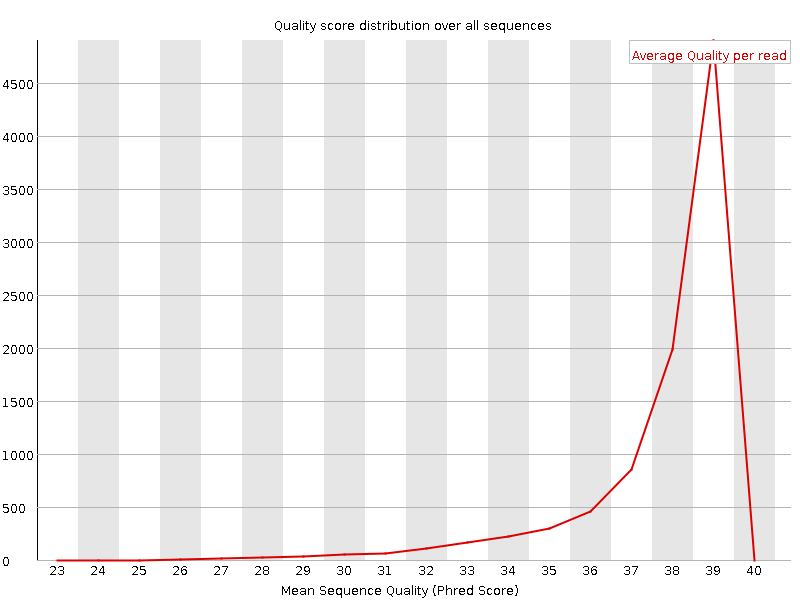
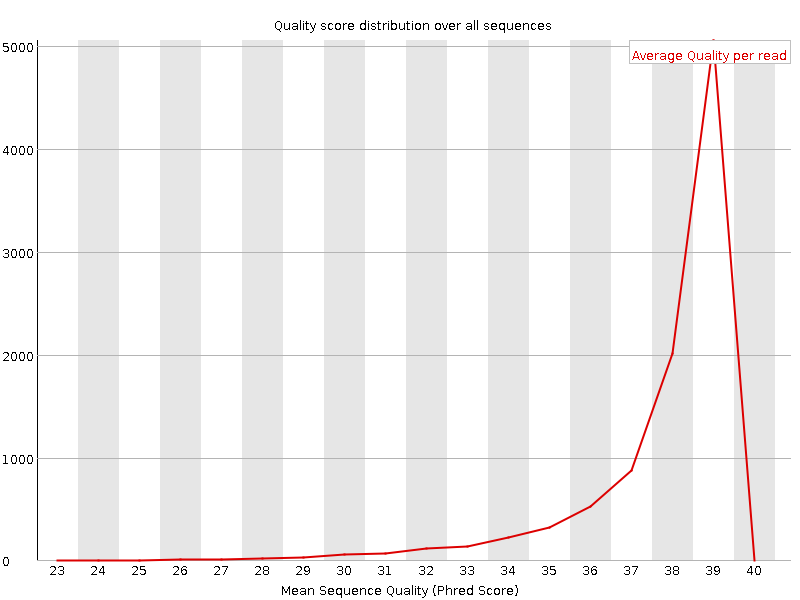
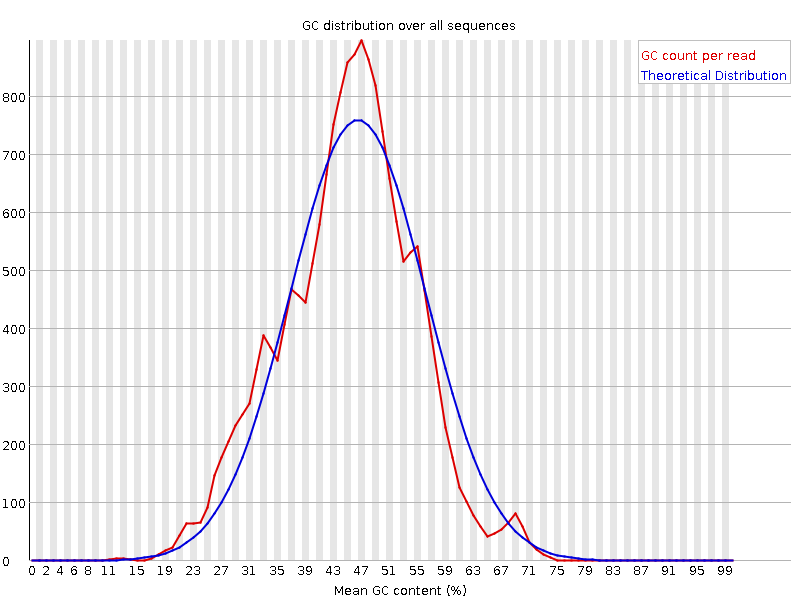
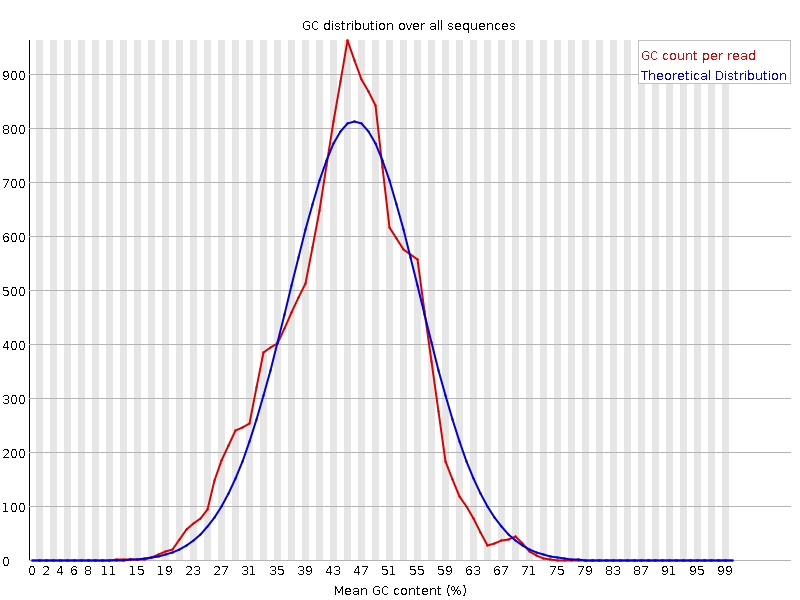
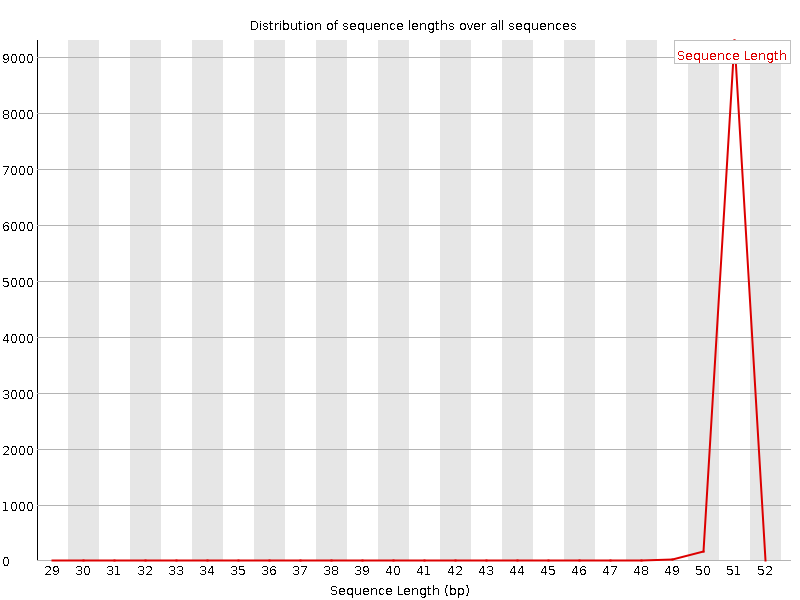
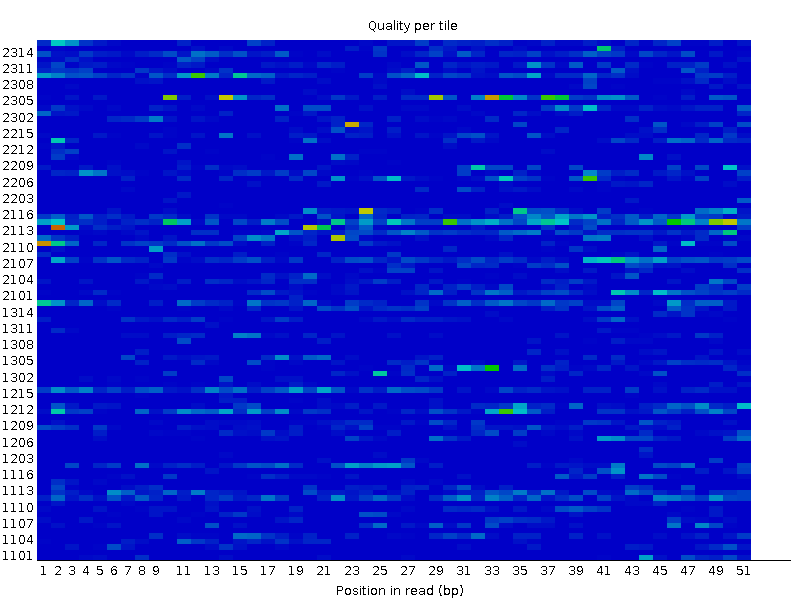
Анализ прочтений был осуществлен с помощью прогрммы FASTQC, установленной на сервере kodomo. Результатом работы программы является папка, содержащая отчет о качестве прочтений, сопровожденный различными графическими изображениями, ознакомится с которыми можно ниже (Таблица 1., Рис.1-14). Программа была запущена с помощью следующей команды (приведены две команды для двух разных реплик): | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| fastqc chr16.1.fastqfastqc chr16.2.fastq | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Таблица 1. Параматры качества ридов для двух реплик.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
КАРТИРОВАНИЕ ЧТЕНИЙ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Картирование чтений проводилось с помощью программы Hisat2 в несколько этапов: 1.Экспорт программы в текущую директорию | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2.Индексирование референсной последовательности (было произведено ранее в 11 практикуме) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| hisat2-build chr16.fasta chr16 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3.Построение выравнивания прочтений и референса в формате .sam (был убран параметр --no-spliced-alignment т.к. работы ведется с транскриптомом, последовательностями с уже вырезанными после сплайсинга интронами. Таким образом риды могут содержать разрывы относительно референсного генома.) Параметр --no-softclip запрещает отрезать нуклеотиды. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| hisat2 -x chr16 -U chr16.1.fastq --no-softclip > alignment_chr16.1.sam hisat2 -x chr16 -U chr16.2.fastq --no-softclip > alignment_chr16.2.sam | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Выход программы Hisat2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Информация о работе программы Hisat2 для обеих реплик приведена в Таблице 2. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Таблица 2. Выход программы Hisat2.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
АНАЛИЗ ВЫРАВНИВАНИЯ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1. Полученный файл с выравниванием был переведен в бинарный формат .bam c использованием программы из пакета samtools - samtools view: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| samtools view alignment_chr16.1.sam -bo alignment_chr16.1.bam samtools view alignment_chr16.2.sam -bo alignment_chr16.2.bam | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2. Выравнивание чтений с референсом было отсортировано по координате в референсе начала чтения при помощи программы samtools sort: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| samtools sort alignment_chr16.1.bam -T temporary.txt -o sort.chr16.1.bam samtools sort alignment_chr16.2.bam -T temporary.txt -o sort.chr16.2.bam | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3. Командой samtools index полученный файл был проиндексирован: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| samtools index sort.chr16.1.bamsamtools index sort.chr16.1.bam | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Определение среднего значения покрытия экзона чтениями | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1. С помощью команды samtools depth было вычислено покрытие чтениями для каждого нуклеотида. С помощью функции МАКС из пакета Excel было найдено максимальное значение покрытия - 247 для нуклеотидов 24227254-24227260 реплики 1 и знаечение покрытия - 9 для 682 двух нуклеотидов. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| samtools depth sort.chr16.1.bam>sort.chr16.1.txtsamtools depth sort.chr16.1.bam>sort.chr16.1.txt | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2. С использованием GenomeBrowser для 16 хромосомы версии Hg19, был обнаружен экзон в состав которого входит данные нуклеотиды реплики 1 и участок гена, на котором расположен один из нуклеотидов с покрытием 9 (23849519) второй реплики, и координаты этих участков. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Рис.15 Ген и Экзоны в состав которых входят нуклеотиды 24227254-24227260 (реплика 1). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Рис.16 Ген в состав которого входит нуклеотид 23849519 (реплика 2).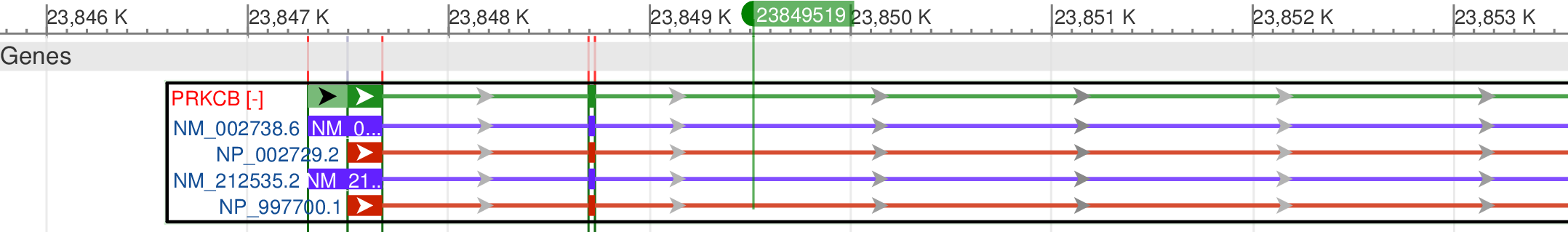 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3. Для каждого нуклеотида входящего в экзон реплики 1 вновь были посчитаны покрытия. С помощью функции СРЗНАЧ пакета Excel было посчитано среднне покрытие для данного экзона, которое составило 1,2. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| samtools depth -r chr16:23,847,300..24,231,932 sort.chr16.1.bam>exon1.txt | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Как видно из приведенных выше результатов, покрытие первой реплики существенно выше, чем покрытие второй (максимальное значение покрытий второй реплики всего 9). Однако несмотря на присутствие высоких значений покрытий в первой реплики, среднее ее покрытие довольно низкое (1,2; среднее покрытие второй реплики - 0,56). В определенном для реплики 1 экзоне, содержащим множество нуклеотидов с очень высоким значением покрытия большинство нуклеотидов оказались непокрыми вообще, из-за чего среднее значение покрытия экзона оказалось довольно низким. Нуклеотиды с наибольшим покрытием второй реплики не попали в границы экзона. В целом покрытие для двух данных реплик довольно низкое. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
ПОДСЧЕТ ЧТЕНИЙ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1. Для подсчета чтений использовалась программа bedtools. Сначала все необходимое для работы программы было экспортировано в нужную директорию с помощью команды: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| export PATH=${PATH}:/P/y14/term3/block4/SNP/bedtools2/bin/ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2. Затем полученные ранее файлы с выравниваниями были конвертированы в формат .bed, удобный для работы программы: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| bedtools bamtobed -i sort.chr16.1.bam>sort.chr16.1.bedbedtools bamtobed -i sort.chr16.2.bam>sort.chr16.2.bed | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3. После этого была использована программа bedtools intersect: парамаетр -a задает файл с рефернсной последовательностью (в данном случае разметка человеческого генома по версии Gencode для генома человека сборки hg19), параметр -b задает файл с последовательностью для пересечения с референсом (файл с выравниванием), парметр -с подсчитывает число ридов, пересекшихся с данным фрагментом разметки. Таким образом на выходе получен файл с координатами пересечений ридов с генами хромосомы 16, названиями генов и числом ридов, образовавших данные пересечения. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| bedtools intersect -a gencode.genes.bed -b sort.chr16.1.bed -c>overlap.1.bedbedtools intersect -a gencode.genes.bed -b sort.chr16.2.bed -c>overlap.2.bed | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
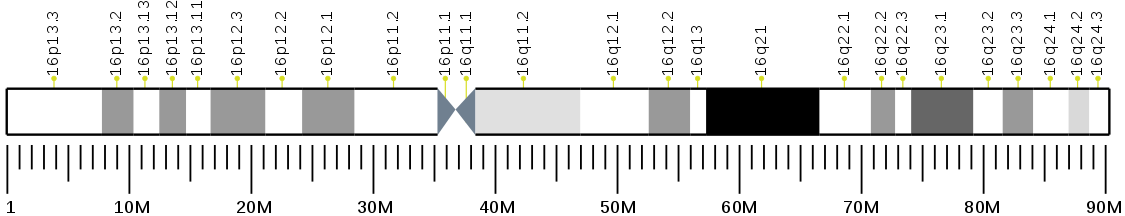
В результате анализа файла, полученного для первой реплики было выявлено, что файл с выравниванием пересекается лишь с одним геном 16 хромомсомы - PRKCB. При этом с разметкой данного гена из файла пересекается лишь 9121 ридов из 9286. В ходе анализа возникли некоторые сложности с подсчетом, так как фрагменты гена, обозначенные в разметке перекрывались сами с собой или были дублированы из-за чего один рид мог попадать сразу в несколько участков, поэтому суммирование по столбцу ридов перекрывющихся с референсом давало значение в 10 раз превышающее число ридов из файла. Однако первая же позиция из файла для данного гена по координатам содержала в себе все остальные координаты, поэтому было взято это значение. Аналогичная ситуация наблюдалась и для второй реплики. Реплика перекрывалась лишь с одним, тем же самым геном разметки PRKCB. Число перекрывающихя ридов было подсчитано аналогичным способом и составило 9356 из 9517. Ниже, в Таблице 3,4 и на Рис.17 приведена информация о разметке гена PRKCB, полученная из NCBI.[4] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Таблица 3. Информация о гене PRKCB.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Рис.17 Схема гена PRKCB. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Таблица 4. Разметка гена PRKCB (координаты гена - 23,847,300..24,231,932)
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Функция PRKCB | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
PRKCB (белковая киназа C Beta) является белок-кодирующим геном. Протеинкиназа C (PKC) - семейство серин- и треонинспецифических протеинкиназ, которые могут быть активированы кальцием и диацилглицеролом. Члены семейства PKC фосфорилируют широкий спектр белковых мишеней и, как известно, участвуют в различных клеточных сигнальных путях. Также белки данного семейства служат основными рецепторами эфиров форбола, класса промоторов опухолей. Протеинкиназа С beta (PRKCB) участвует осуществляет множество функций, таких как активация B-клеток, индукция апоптоза, пролиферация эндотелиальных клеток и поглощение сахара в кишечнике. Заболевания, связанные с PRKCB, включают глиобластому и гипергликемию.Среди его метаболических путей - GABAergic синапс и Common Cytokine Receptor Gamma-Chain Family Signaling Pathways.[5] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
BEDTOOLS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
1. BAM -> FASTQ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
С помощью подпрограммы bedtools - bamtofastq файл в формате .bam может быть переведен в формат .fq. Таким образом, с помощью следующей команды файлы с выравниваниями в формате .bam были переведены в формат fastq: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| bedtools bamtofastq -i sort.chr16.1.bam -fq sort.chr16.1.fqbedtools bamtofastq -i sort.chr16.2.bam -fq sort.chr16.2.fq | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
2. FASTA ФАЙЛ ДЛЯ ГЕНА PRKCB | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| С помощью программы bedtools ранее были обнаружены пересечения выравнивания с разметкой генома - информация об этом приведена выше. Из файла, содержащего информацию о координатах перессечения разметки с выравниванием, полученного следующим образом: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| bedtools intersect -a gencode.genes.bed -b sort.chr16.1.bed -u>overlap.chr16.1.bed | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Где, -u - параметр, определяющий выдачу координат пересечения лишь один раз, если A пересеклось с B один и более раз (не выдает информацию о количетсве ридов, попавших в пересечение, содержит лишь пересечения в которые попало 1 и более ридов, выдает их лишь по одному разу), был взята только первая строчка, содержащая координаты гена. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Далее с помощью программы bedtools getfasta из fasta файла со всей 16 хромосомы была получена последовательность самого гена PRKCB и записана в файл 1.fasta: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| bedtools getfasta -fi chr16.fasta -bed overlap.chr16.1.bed>1.fasta | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3. РАЗБИЕНИЕ ХРОМОСОМЫ НА ФРАГМЕНТЫ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Для того чтобы разбить хромосому на фрагменты использовалась подпрограмма bedtools makewindows. Для ее использования применялся параметр -g, требующий в качетсве входного файла файл, содержащий название хромосому и ее длину. Согласно данным c NCBI длина 16 хромосомы для сборки hg19 - 90354753bp, что и было записано в файл chr16.txt. Далее, с помощью следующей команды длина хромосомы была поделена на 91 интервал длины 1 000 000bp: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| bedtools makewindows -g chr16.txt -w 1000000 >chr16_intervals.txt | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
4. ОБЪЕДИНЕНИЕ ЧТЕНИЙ В КЛАСТЕРЫ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Объединение чтений в кластеры производилось с помощью программы bedtools cluster. На вход программе был дан полученный ранее файл с выравниванием в формате .bed. Параметр -d задает максимальное расстояние на котором интервалы могут объединятся в кластеры. На выход был получен отсортированный файл с дополнительным столбцом. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| bedtools cluster -i sort.chr16.1.bed -d 10 >sort.chr16.1.clusters10.bed | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
5. СОЗДАНИЕ СЛУЧАЙНЫХ ИНТЕРВАЛОВ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
C помощью программы bedtools random были созданы 1000 случайных интервалов (-n) длинной 200 bp (-l). Для этого на вход программе был дан созданный ранее файл chr16.txt, содержащий информацию о длине хромосомы 16. Полученный фаул содержал координаты случайных интевалов, их длинну, цепь и нумерацию. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| bedtools random -l 200 -n 1000 -g chr16.txt >chr16.random.txt | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
6. КООРДИНАТЫ 3' ОБЛАСТИ PRKCB | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Координаты 3' области гена PRKCB были получены с помощью подпрограммы flank. Для этого использовался параметр -r, задающий отступ от координаты конца исходного интервала. На вход давался файл с размером хромосомы и еще один, с известными координатами гена. На выход был получен файл с измененными координатами. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| bedtools flank -l 0 -r 1000 -g chr16.txt -i overlap.chr16.PRKCB.bed >flank.txt | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
7. РАСШИРЕННЫЕ В ОБЕ СТОРОНЫ КООРДИНАТЫ ГЕНА | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Для того, чтобы расширить в обе стороны координаты гена PRKCB использовалась подпрограмма slop c праметром -b, осуществлюещим расширение именно в обе стороны на заданную длину. На выход получен файл гена с измененными координатами. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| $bedtools slop -b 1000 -g chr16.txt -i overlap.chr16.PRKCB.bed >slop.txt | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
8. СДВИНУТЫЕ КООРДИНАТЫ ГЕНА | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Для того, чтобы получить сдвинутые к началу хромомсомы координаты гена использовалась программы shift с параметром -s, который обеспечивает сдвиг и начальной, и конечной координаты гена. Однако по умолчанию координаты сдвигаются в сторону направления конца хромосомы, поэтомы значение 500 было взято со знаком -. Любоптытно также, у данной программы существуют параметры, позволяющие задавать разные параметры сдвига для +/- цепей (-p, -m). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| bedtools shift -s -500 -g chr16.txt -i overlap.chr16.PRKCB.bed >shift.txt | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
9. НЕПЕРЕСЕКАЮЩАЯСЯ ФРАГМЕНТЫ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Для того, чтобы получить фрагменты хромосмы, не пересекающиеся с ридами была использована программы complement. На вход были даны кординаты хромосомы и выровненный файл с прочтениями, еще не пересеченный с геномом. Можно ознакомится с выдачей программы complement.txt. Из этого файла хорошо видно, что часть ридов не легла в пределы гена PRKCB, но при этом не была определена разметкой, как входящая в состав других геном, а также часть гена оказалась непокрыта ридами, что согласуется с выдачей программы samtools depth. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| bedtools complement -g chr16.txt -i sort.chr16.1.bed >complement.txt | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
10.ПОКРЫТИЕ РИДАМИ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Покрытие для каждого интервала пересечения ридов с разметкой генома было посчитано с помощью подпрограммы genomecov. На выходе программы получился файл, содержащий координаты интервалов и их покрытие ридами в следующей колонке. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| bedtools genomecov -g chr16.txt -i overlap.chr16.1.bed -bg>coverage.txt | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
КОМАНДЫ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Таблица 5. Команды, использованные в данном практикуме.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
ССЫЛКИ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||