Практикум 11. Работа с чтениями хромосомы человека.
Анализ качества чтений
Для анализа качества прочтений, лежащих в файле chr11.fastq, из рабочей директории была запущена программа fastqc:
fastqc chr11.fastq
Очистка чтений
Для очистки чтений была запущена программа trimmomatic с действиями LEADING:20 (отрезает нуклеотиды с 5' конца, если их качество ниже 20), TRAILING:20 (то же самое для нуклеотидов с 3' конца), и MINLEN:50 (удаляет риды короче 50 нуклеотидов).
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr11.fastq chr11_trimm.fastq LEADING:20 TRAILING:20 MINLEN:50
Результат чистки
1. Число ридов сократилось с 4198 до 4064.
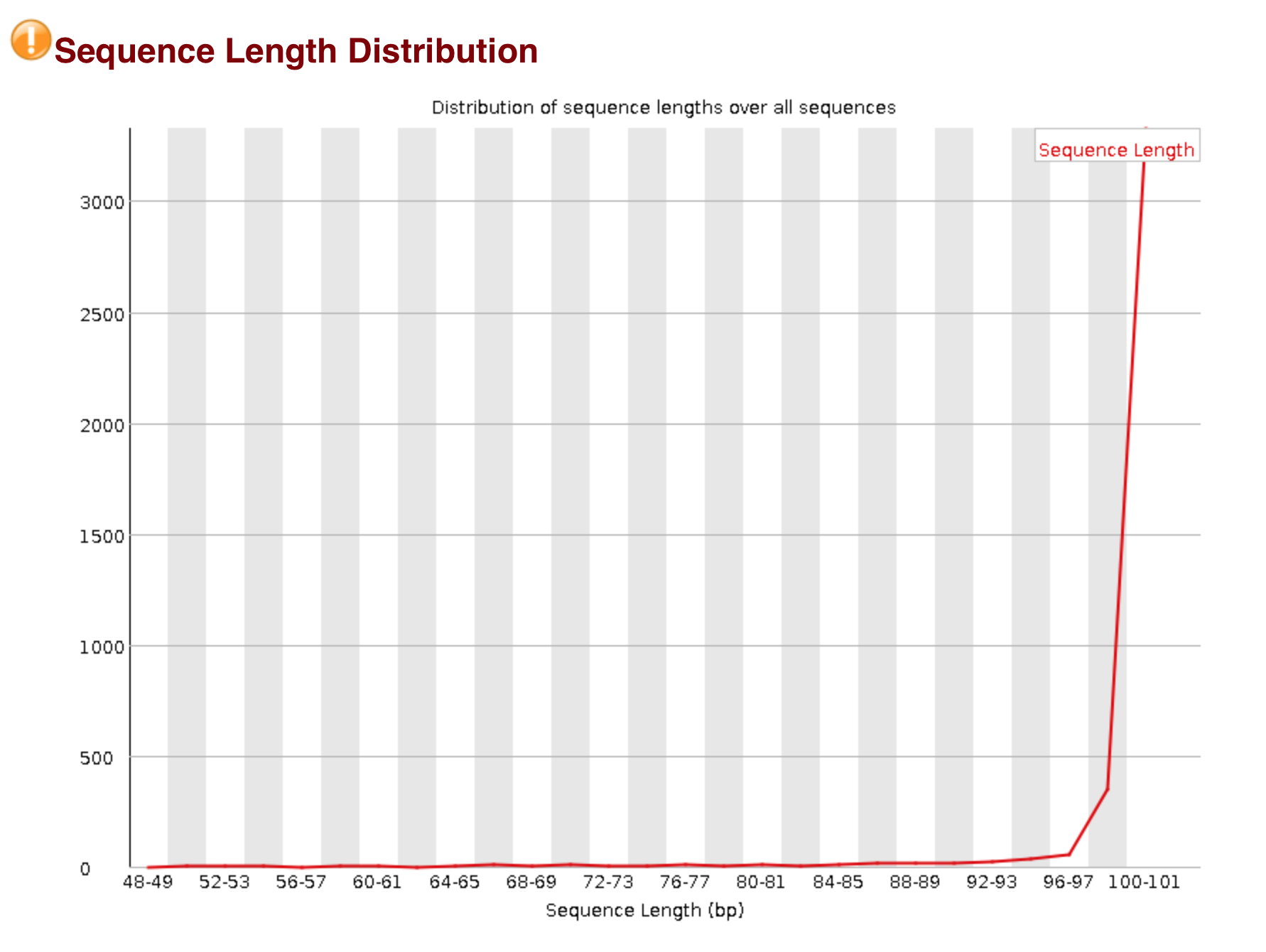
2. Изменилось распределение длины ридов, поскольку у ридов с некачественными концами эти концы обрезались. Иллюстрация приведена на картинках:
Рисунок 1. Распределение длин до trimmomatic
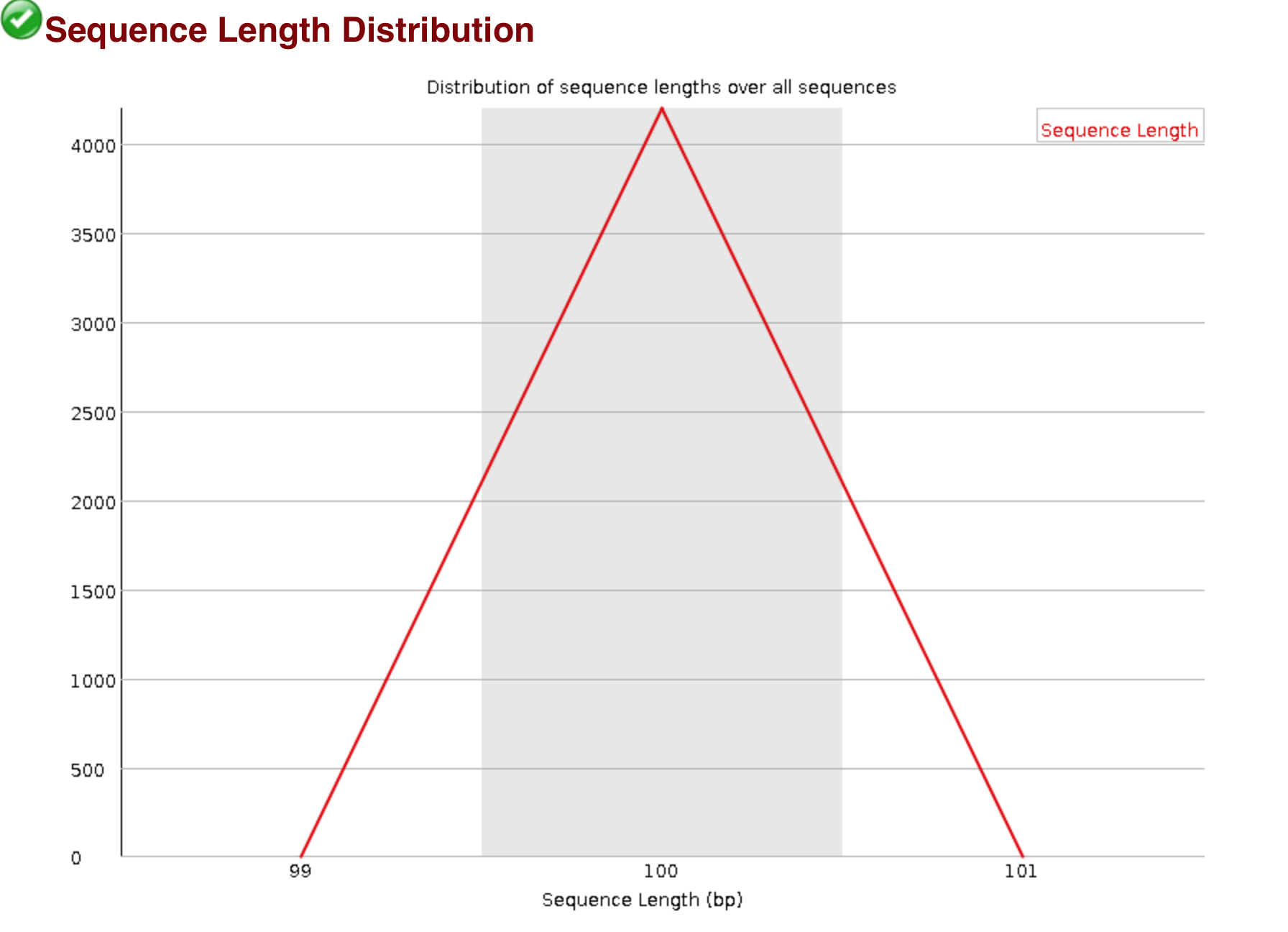
Рисунок 2. Распределение длин после trimmomatic
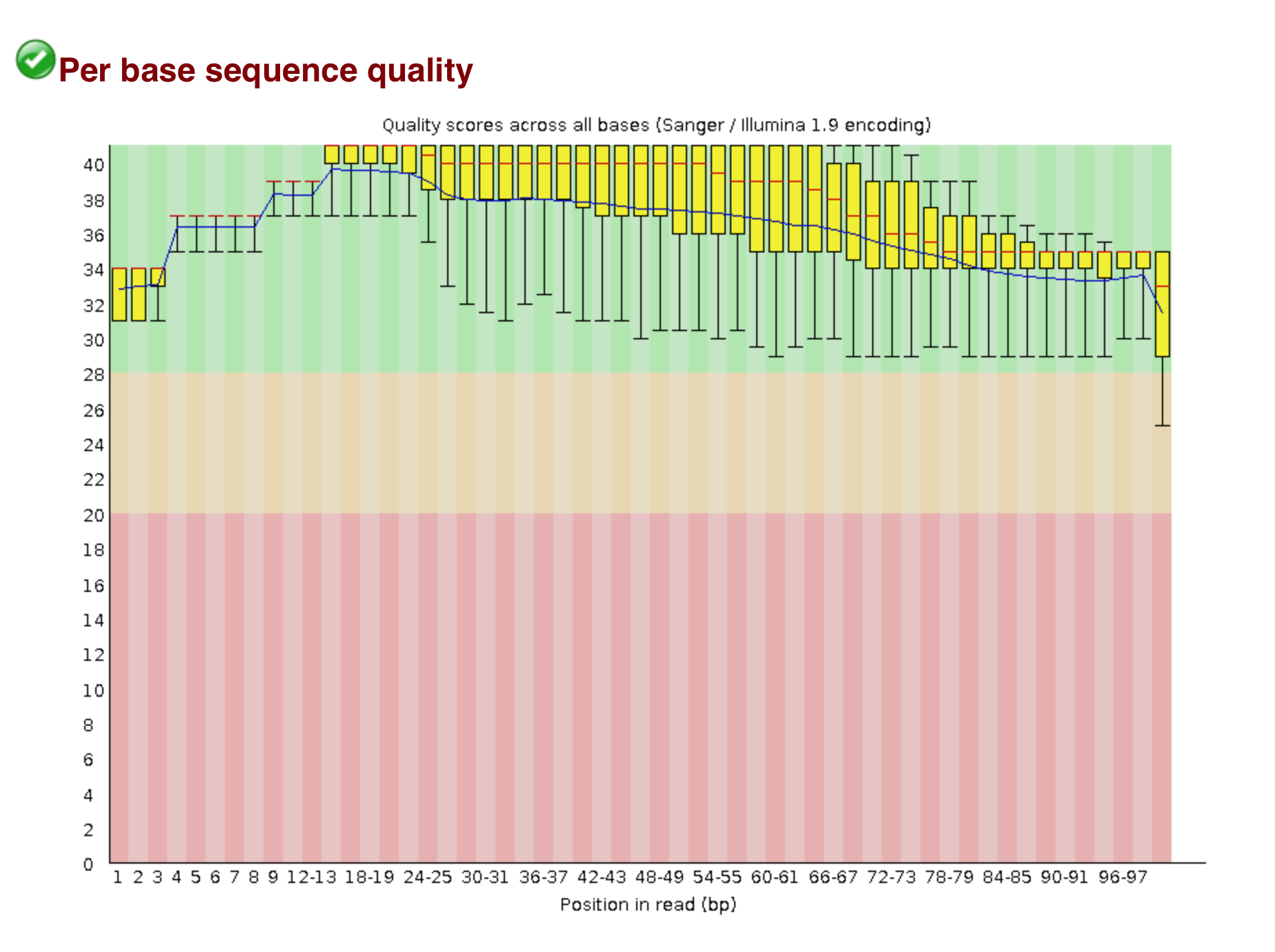
3. Существенно улучшилось среднее качество нуклеотидов, близких к концам, так как некачественные концы вырезались из ридов.
Рисунок 3. Понуклеотидное среднее качество ридов до trimmomatic
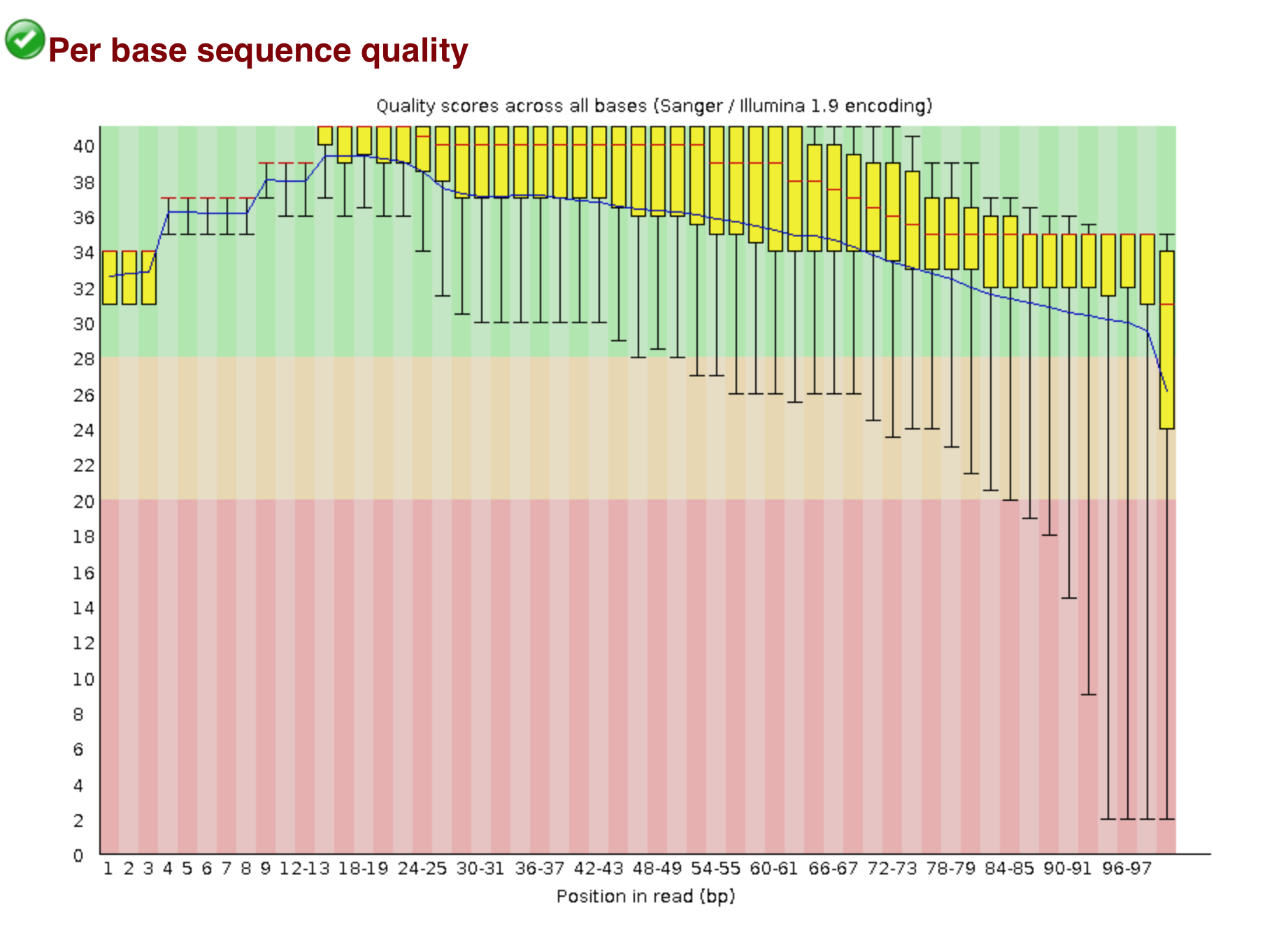
Рисунок 4. Понуклеотидное среднее качество ридов после trimmomatic
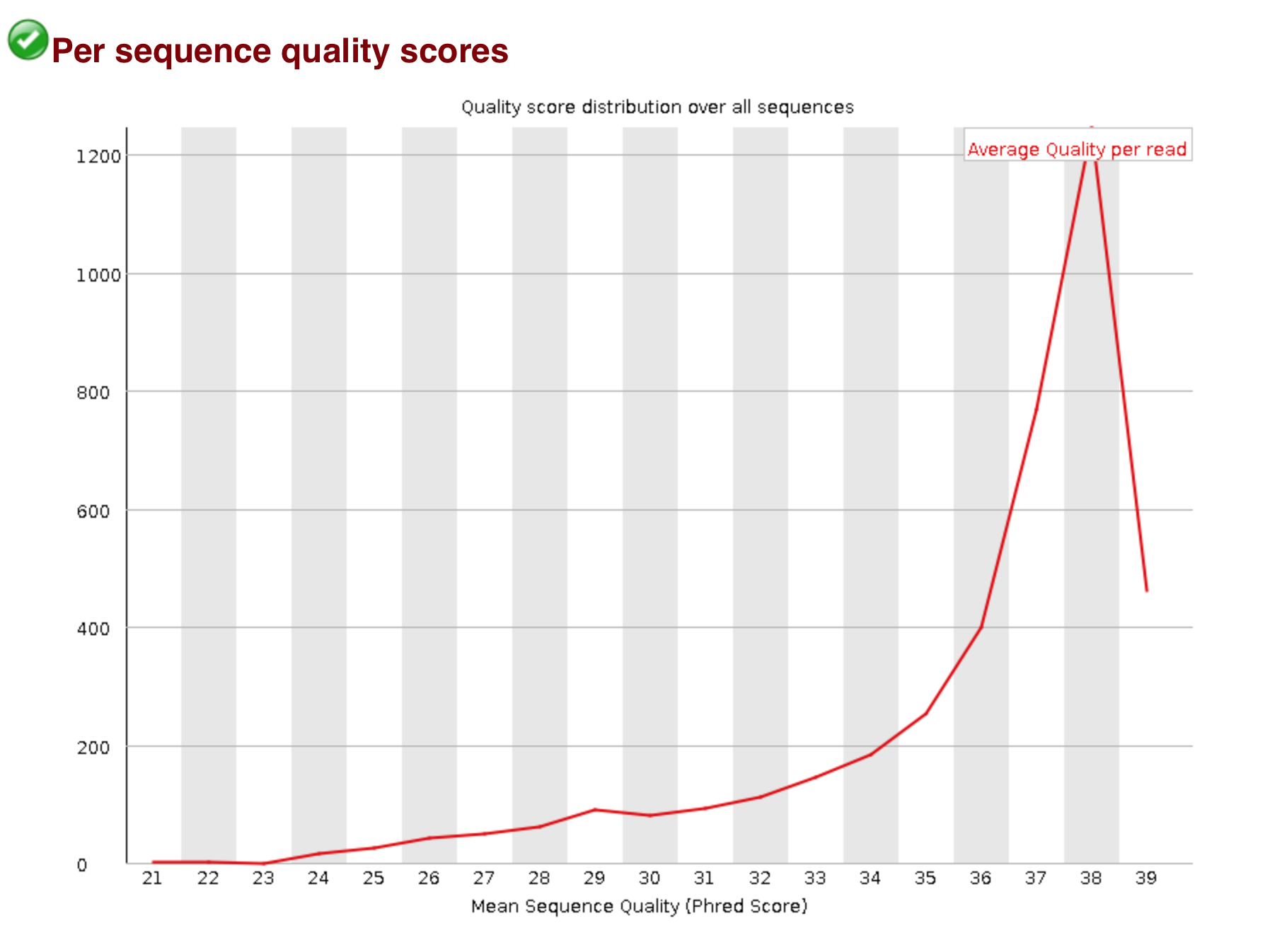
4. Изменилось распределение качества ридов (Phred score). Пик не сместился, но увеличилось количество ридов, близких к нему (ширина пика) и ликвидировались риды совсем плохого качества (после чистки качество > 21)
Рисунок 5. Распределение phred score до trimmomatic
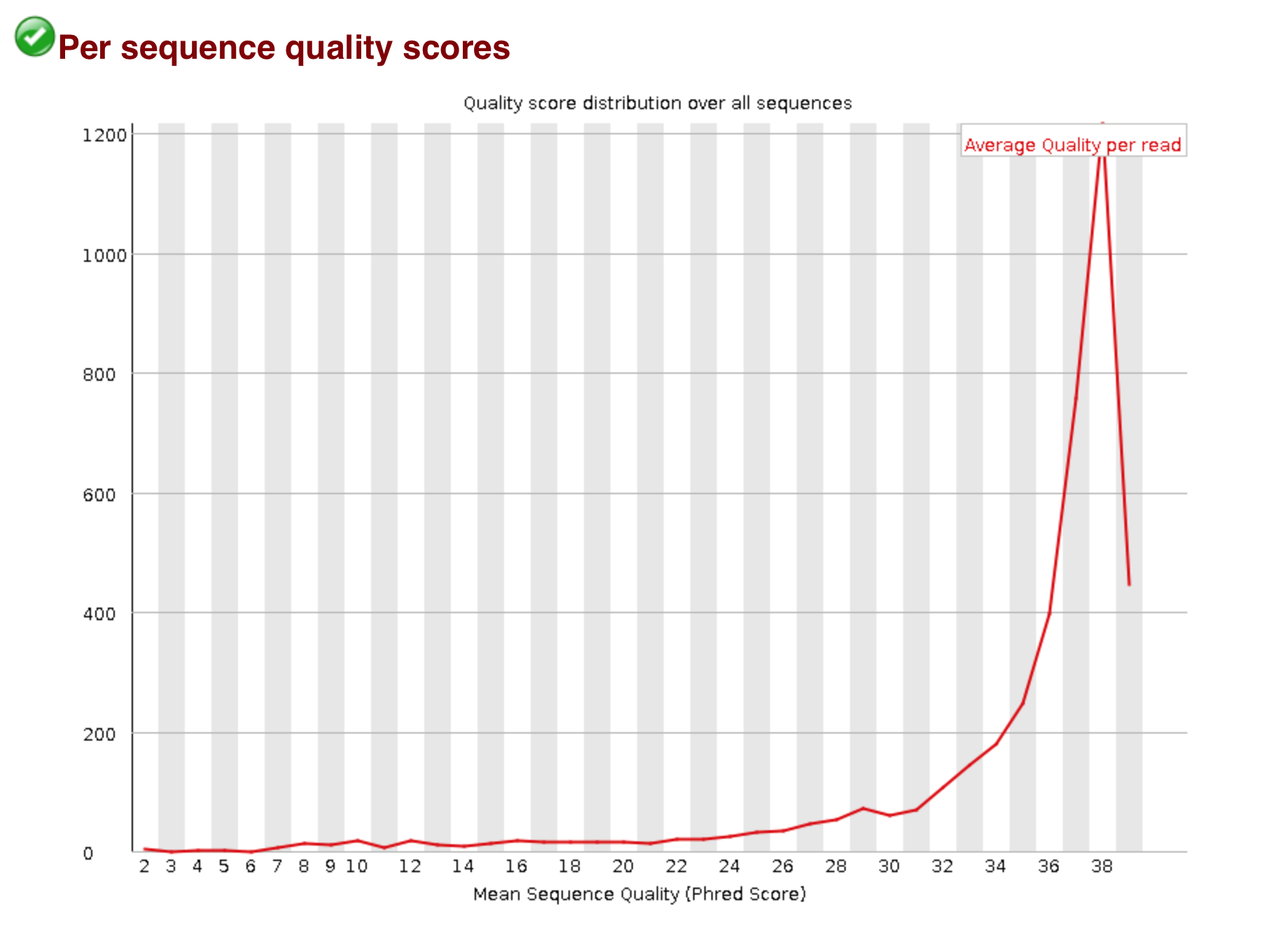
Рисунок 6. Распределение phred score после trimmomatic
Картирование чтений
Картирование чтений - это по сути создание выравнивания чтений и референсной последовательности
Команды:
# Индексируем референсную последовательность
hisat2-build chr11.fasta chr11_index
# Строим выравнивание ридов и референса
hisat2 -x chr11_index -U chr11_trimm.fastq -S chr11_align_ref_reads.sam --no-spliced-alignment --no-softclip
4064 reads; of these:
4064 (100.00%) were unpaired; of these:
3 (0.32%) aligned 0 times
4051 (99.68%) aligned exactly 1 time
0 (0.00%) aligned >1 times
99.68% overall alignment rate
Из выдачи программы hisat2 узнаем, что из 4064 ридов на нашу хромосому было картировано 4051 ридов (99.68%), а 13 ридов (0.32%) картировано не было.
Преобразования выравнивания чтений и референса
Команды:
# Переводим выравнивание в бинарный формат (.bam), так как он удобнее для компьютера
samtools view -b chr11_align_ref_reads.sam >> chr11_align_ref_reads.bam
# Сортируем выравнивание по координате в референсе начала рида
samtools sort chr11_align_ref_reads.bam chr11_align_sorted
# Индексируем отсортированный .bam файл
samtools index chr11_align_sorted.bam
Поиск SNP и инделей
Команды:
# Создаем файл с полиморфизмами
-o -- output to FILE
samtools mpileup -uf chr11.fasta -o polym.bcf chr11_align_sorted.bam
# Создаем файл со списком отличий между ридами и референсной последовательностью
bcftools call -cv -o differ.vcf polym.bcf
| координаты полиморфизма |
тип полиморфизма |
было в референсе |
найдено в чтениях |
глубина покрытия данного места |
качество чтений в данном месте |
| chr11: 17408305 |
замена |
G |
C |
14 |
103.008 |
| chr11:17408404 |
замена |
C |
T |
12 |
56.0072 |
| chr11: 17408630 |
замена |
C |
T |
35 |
221.999 |
Из файла differ.vcf видим, что было найдено 23 полиморфизма, из которых 2 - индели, остальные - snp. Глубина покрытия от 1 до 92. При таком разбросе данных считать среднее значение на мой взгляд не имеет смысла. Качество прочтения тоже разнится сильно: от 4.76921 до 225.009.
Аннотация SNP
Проаннотируем найденные snp по 5 базам данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar
Команды:
# Файл со списком отличий между ридами и референсной последовательностью переводим в формат, с которым умеет работать annovar
convert2annovar.pl -format vcf4 differ.vcf > chr11.avinput
NOTICE: Finished reading 55 lines from VCF file
NOTICE: A total of 23 locus in VCF file passed QC threshold, representing 21 SNPs (17 transitions and 4 transversions) and 2 indels/substitutions
NOTICE: Finished writing 21 SNP genotypes (17 transitions and 4 transversions) and 2 indels/substitutions for 1 sample
Приступаем к непосредственной аннотации
#Аннотация по базе refgene
annotate_variation.pl -out refgen -build hg19 chr11.avinput /nfs/srv/databases/annovar/humandb.old/
Из файла refgen.variant_function видим, что база данных Refseq делит snp по их локализации (exonic - 5, intronic - 14, UTR3 - 4). Из этого же файла видим, в какие гены попали наши snp: KCNJ11, BUD13, ZPR1.
#Аннотация по базе dbsnp
annotate_variation.pl -filter -out SR_SNP -build hg19 -dbtype snp138 chr11.avinput /nfs/srv/databases/annovar/humandb.old/
Программа показала, что среди наших полиморфизмов есть 19 rs, то есть аннотированных.
#Аннотация по базе 1000 genomes
annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out 1000Genomes chr11.avinput /nfs/srv/databases/annovar/humandb.old/
Аннотация 1000Genomes показывает частоты аллелей в output - файле. Максимальная частота - 0.737021 (17409572 позиция, T/C), минимальная - 0.00339457 (17408305 позиция, G/C). В нашем случае аннотация дала результат для 19 полиморфизмов. Для каждого из них также указано,
относится он к het или к homo, то есть во всех ли наших ридах встречается один и тот же ваиант.
#Аннотация по базе GWAS
annotate_variation.pl -regionanno -build hg19 -out GWAS -dbtype gwasCatalog chr11.avinput /nfs/srv/databases/annovar/humandb.old/
Программа показала, что некоторые из наших snp (а именно 6 штук) связаны с возникновением заболеваний: type 2 diabetes, triglycerides (что это такое непонятно), Triglycerides-Blood Pressure (TG-BP)б Metabolic syndrome (bivariate traits), Obesity-related traits
#Аннотация по базе Clinvar
annotate_variation.pl chr11.avinput -filter -dbtype clinvar_20150629 -buildver hg19 -out CLINVAR /nfs/srv/databases/annovar/humandb.old/
В базе ClinVar содержится информация о том, какое влияние оказывают полиморфизмы на здоровье человека. Программа не дала какой-либо клинической информации о наших полиморфизмах.
Каждая команда генерирует файл .log, который копирует ее же ответ через консоль (комментарии по ходу работы программы), а также 2 файла с непосредственным результатом.
Поизучаем немного обнаруженные snp. Мутации по chr11:17408630 и chr11:17409572 приходятся на ген KCNJ11 (кодирует калиевый канал). Из аннотации по GWAS видим, с каким забоолеванием связана конкретная snp. Упомянутые выше мутации ответственны за развитие диабета второго типа. Результат вполне объясним с биологической точки зрения: калий и глюкоза под действием инсулина всасываются в клетку симпортно, поэтому анормальная работа калиевого канала может вызывать нарушения баланса глюкозы.
P.S. Все файлы, полученные в ходе выполнения практикума, Вы можете найти в директории srv/databases/ngs/anrozina