Практикум 12. Картирование чтений, полученных в результате секвенирования транскриптома
Резюмирующая таблица команд
| номер задания |
команда |
функция |
| 1 |
fastqc chr11.1.fastq |
анализ качества чтений |
| 2 |
hisat2 -x ../chr11_index -U chr11.1.fastq -S chr11_align_ref_reads.sam --no-softclip |
Картирование чтений на референс |
| 3 |
samtools view -b chr11_align.sam >> chr11_align.bam |
Перевод выравнивания в бинарный формат |
| 3 |
samtools sort chr11_align.bam chr11_align_sorted |
Сортировка выравнивания по координате начала рида в референсе |
| 3 |
samtools index chr11_align_sorted.bam |
Индексирование отсортированного .bam файла |
| 4 |
htseq-count -f bam -s yes -i gene_id -m union chr11_align_sorted.bam /P/y14/term3/block4/SNP/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf > count_reads.txt |
Показывает, сколько ридов из выравнивания в формате bam (можно также использовать sam легло на каждый feature (в нашем случае ген) из feature file. |
| 5 |
python count.py count_reads.txt |
Запуск скрипта, который выдает строки с feaures, на которые пришлось больше чем ноль ридов |
1. Анализ качества чтений, чистка
Команды:
fastqc chr11.1.fastq
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr11.1.fastq chr11.1_trimm.fastq LEADING:20 TRAILING:20 MINLEN:50
fastqc chr11.1_trimm.fastq
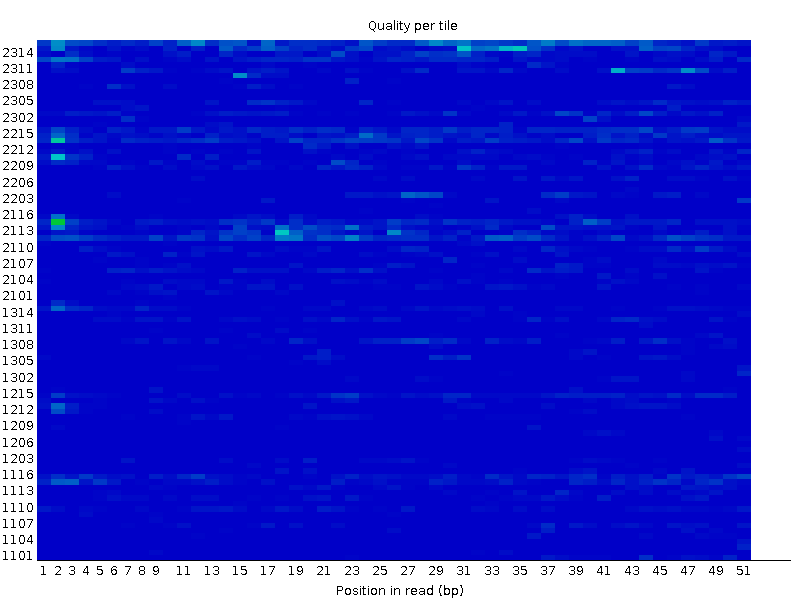
Качество чтений:
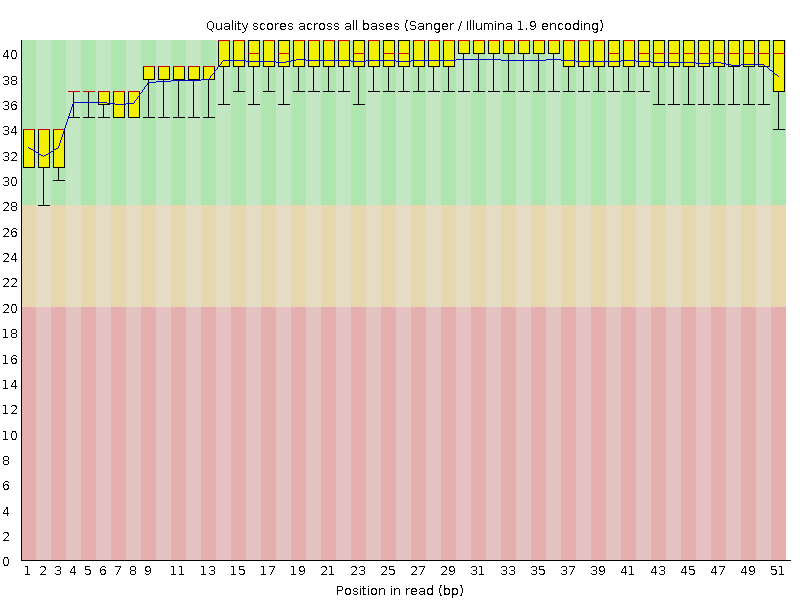
Качество per tile после trimmomatic
2. Картирование чтений
В качестве референсной последовательности берем уже проиндексированную в практикуме 11 хромосому (chr11_index.fasta). При запуске hisat2 убираем параметр --no-spliced-alignment, так как работаем с транскриптомом и должны учитывать сплайсинг.
Команды:
hisat2 -x ../chr11_index -U chr11.1_trimm.fastq -S chr11_align.sam --no-softclip
39103 reads; of these:
39103 (100.00%) were unpaired; of these:
276 (0.71%) aligned 0 times
38827 (99.29%) aligned exactly 1 time
0 (0.00%) aligned >1 times
99.29% overall alignment rate
3. Анализ выравнивания
Команды:
samtools view -b chr11_align.sam >> chr11_align.bam
samtools sort chr11_align.bam chr11_align_sorted
samtools index chr11_align_sorted.bam
4. Подсчет чтений
Команды:
htseq-count -f bam -s yes -i gene_id -m union chr11_align_sorted.bam /P/y14/term3/block4/SNP/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf > count_reads.txt
Функции параметров:
-f тип файла с выравниванием (sam или bam)
-s из цепь-специфичного ли анализа получены данные.
-i gff атрибут, который будет использоваться как feature ID. Будем использовать gene_id
-m модуль для обработки ридов, имеющих больше одной feature. Было решено использовать union, чтобы обнаружить как можно больше перекрываний (режим union более чувствительный в спорных случаях, см. Рисунок 1)
Результат:
__no_feature 38799
__ambiguous 0
__too_low_aQual 0
__not_aligned 276
__alignment_not_unique 0
Рисунок 1. Режимы модуля -m программы htseq-count
![]()
5. Анализ результатов
В задании не уточняется, что должен делать скрипт count.py. Однако из дальнейших формулировок предполагаю, что скрипт count.py должен показывать, на какие гены упали риды и в каком количестве.
Ссылка на скрипт
Команды:
python count.py count_reads.txt
Результат:
['ENSG00000109971.9', '28']
['__no_feature', '38799']
['__not_aligned', '276']
Итак, мы видим, что только 28 ридов легло на ген. Причина не может состоять в том, что для некоторых белков границы генов определены неточно, и транскрипты выходят за те границы, что обазначены в записях генов, и таким образом попадают в категорию no_feature. Ведь при запуске htseq-count для модуля -m мы использовали параметр union. По-видимому, дело в небезупречности работы программы.
Идентефикатор ENSG00000109971 соответствует гену HSPA8 - heat shock protein family A (Hsp70) member 8. Как и другие белки-шапероны, он выполняет функцию фолдинга белков, новосинтезированных и по какой-то причине развернувшихся, а также участвует в деградации мутантных белков.
P.S. Все файлы, полученные в ходе выполнения практикума, Вы можете найти по адресу /srv/databases/ngs/anrozina/pr12
ПЕРЕЗАПУСК
Команды:
htseq-count -f bam -s no -i gene_id -m union chr11_align_sorted.bam /P/y14/term3/block4/SNP/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf > count_reads_1.txt
Функции параметров:
-f тип файла с выравниванием (sam или bam)
-s из цепь-специфичного ли анализа получены данные.
-i gff атрибут, который будет использоваться как feature ID. Будем использовать gene_id
-m модуль для обработки ридов, имеющих больше одной feature. Было решено использовать union, чтобы обнаружить как можно больше перекрываний (режим union более чувствительный в спорных случаях, см. Рисунок 1)
Результат:
__no_feature 137
__ambiguous 43
__too_low_aQual 0
__not_aligned 276
__alignment_not_unique 0
Результат нмного лучше!
5. Анализ результатов
В задании не уточняется, что должен делать скрипт count.py. Однако из дальнейших формулировок предполагаю, что скрипт count.py должен показывать, на какие гены упали риды и в каком количестве.
Ссылка на скрипт
Команды:
python count.py count_reads_1.txt
Результат:
['ENSG00000109971.9', '38640']
['ENSG00000200879.1', '7']
['__no_feature', '137']
['__ambiguous', '43']
['__not_aligned', '276']
Получили более осмысленный результат. Ридов, не попавших на ген, теперь намного меньше тех, что на ген попали.