Практическая работа 12
Задание 1. Программа для сравнения выравниваний
Программа для сравнения двух выравниваний доступна по ссылке.
Задание 2. Сравнение программ MSA
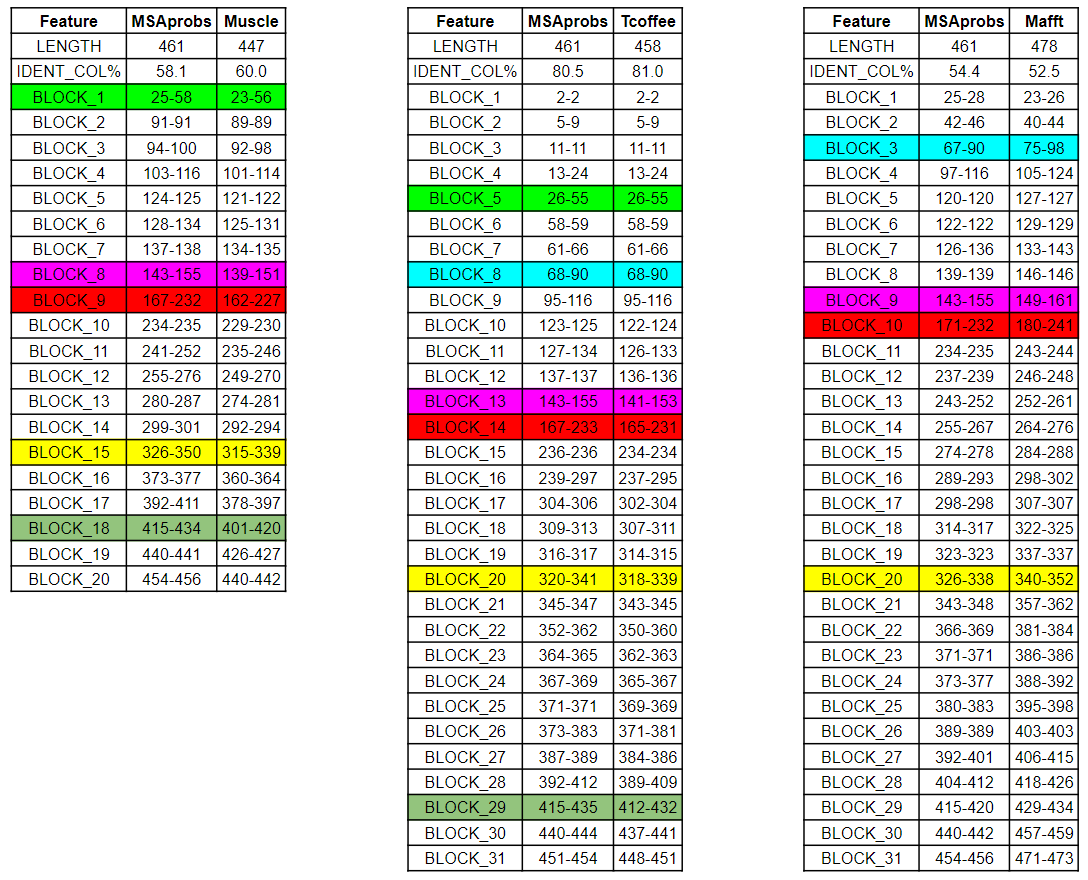
Для выравнивания я выбрал фермент малатдегидрогеназу. Исходный fasta-файл можно скачать по ссылке. Множественное выравнивание осуществялось с помощью 4 программ: MSAprobs(ссылкa), Mafft(ссылкa), Muscle(ссылкa) и Tcoffee(ссылкa). В скобках после названия программы приведена ссылка на соответствующий fasta-файл выравнивания, а проект Jalview со всеми четырьмя выравниваниями можно скачать здесь.
В качестве референсного выравнивания я выбрал выравнивание, построенное программой MSAprobs, результаты сравнения которого с остальными тремя выравниваниями приведены в Табл. 1. Списки одинаково выравненных колонок можно скачать по следующим ссылкам: MSAprobs-Muscle, MSAprobs-Tcoffee, MSAprobs-Mafft. Поскульку каждая программа множественного выравнивания использует свой собственный эвристический алгоритм, то неудивительно, что все четыре варианта выравнивания отличаются. Однако стоит заметить, что все четыре алгоритма практически одинаково выравнили три блока(выделенные розовым, красным и желтым), которые отвечают высококонсервативным участкам фермента. Таким образом, использование разных программ для множественного выравнивания и дальнейшее сравнение их результатов позволяет более точно указать и выявить консервативные участки белка.
Задание 3. Пространственное выравнивание
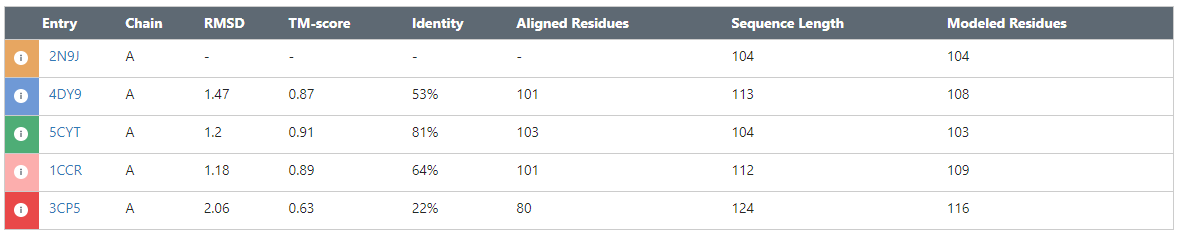
Для построения пространственного выравнивания я выбрал домен Cytochrome c (PF00034) и следующие белки для выравнивания:
- 2N9J (Homo sapiens)
- 4DY9 (Leishmania major)
- 5CYT (Thunnus alalunga)
- 1CCR (Oryza sativa)
- 3CP5 (Rhodothermus marinus)
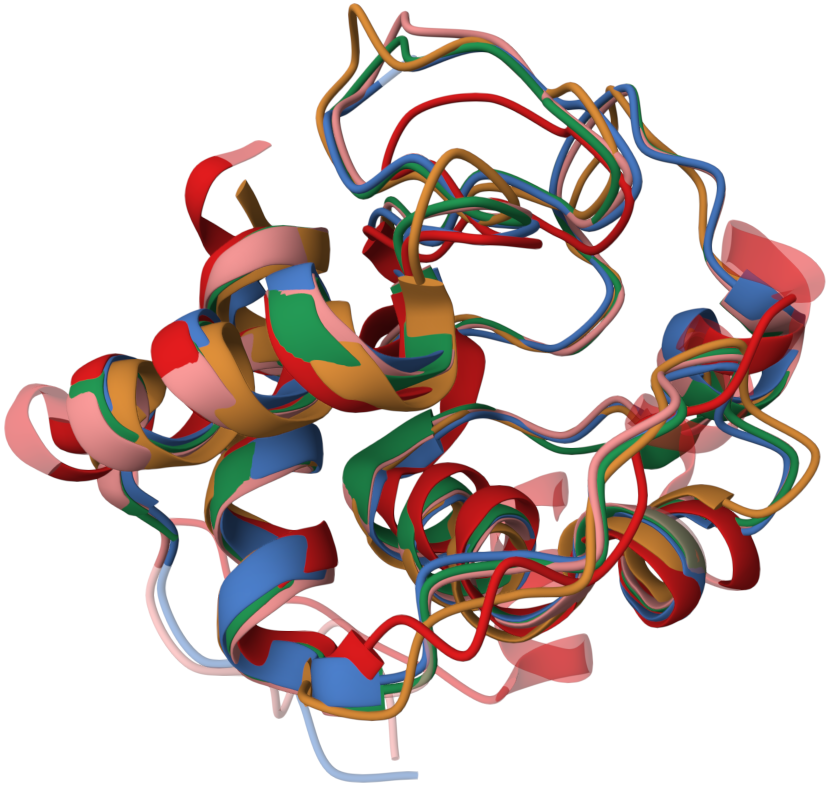
Пространственное выравнивание осуществлялось на сайте PDB по алгоритму TM-align. Полученные результаты представлены в Табл. 2 и на Рис. 1-2. Кроме того, было проведено выравнивание этих же последовательностей с помощью алгоритма обычного множественного выравнивания MSAprobs(см. Рис. 3). Проект Jalview с пространственным выравнивание можно скачать по ссылке, а проект с множественным выравнивание MSAprobs – по ссылке. В целом, и прастранственное выравнивание и выравнивание последовательностей практически одинаковы с точки зрения выявления гомологичных участков. Возможно это связано с тем, что белок цитохром с очень консервативен и медленно изменяется в процессе эволюции, и поэтому простого выравнивания последовательностей достаточно, чтобы выявить гомологию.


Описание программы для множественного выравнивания CLUSTAL
Программа CLUSTAL на первом этапе осуществляет парные выравнивания всевозможных пар последовательностей по алгоритму Вильбура-Липмана. Затем, строится матрица схожести и на ее основе филогенетическое дерево методом UPGMA. Далее берутся две наиболее близкие последовательности и строится их парное выравнивание по Вильбуру-Липману. Теперь этот кластер из двух последовательностей будет представлен одной консенсусной последовательностью, которая получается из выравнивания по следующему алгоритму: 1) совпадающие буквы не изменяются, 2) несовпадающие буквы заменяются на X, 3) гепы остаются на месте. Далее осуществляется выравнивание полученной консенсусной последовательности с другой послеловательность или с другой консенсусной последовательностью, представляющий другой кластер. Последние два шага повторяются до тех пор, пока программа не обойдет все кластеры дерева. Так же есть некоторые способы повысить чувствительность алгоритма, особенно при построении консенсусной последовательности. Подробнее здесь: Higgins, D. G., & Sharp, P. M. (1988). CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene, 73(1), 237–244. doi:10.1016/0378-1119(88)90330-7