Практикум 12. Анализ транскриптомов
Часть I: подготовка чтений
Для выполнения данного практикума использовались прочтения транскриптома с 8-ой хромосомы пациента. Вначале качество ридов было визулизировано программой FastQC. Затем с конца каждого чтения были удалены нуклеотиды с качеством ниже 20, оставлены только чтения длиной не меньше 50 нуклеотидов. Затем качество ридов было снова визуализировано
Таблица с командами (исполнялись в папке /nfs/srv/databases/ngs/anton.vlasov/pr12):
| Команда | Что делает |
| fastqc chr8.1.fastq | Визуализирует качество ридов транскриптома 8-ой хромосомы. |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr8.1.fastq chr8_good.fastq TRAILING:20 MINLEN:50 | Удаляет с конца каждого чтения нуклеотиды с качеством ниже 20, оставляет чтения длиной не меньше 50. |
| fastqc chr8_good.fastq | Визуализирует качество улучшенных ридов транскриптома. |

Per base quality до обработки.
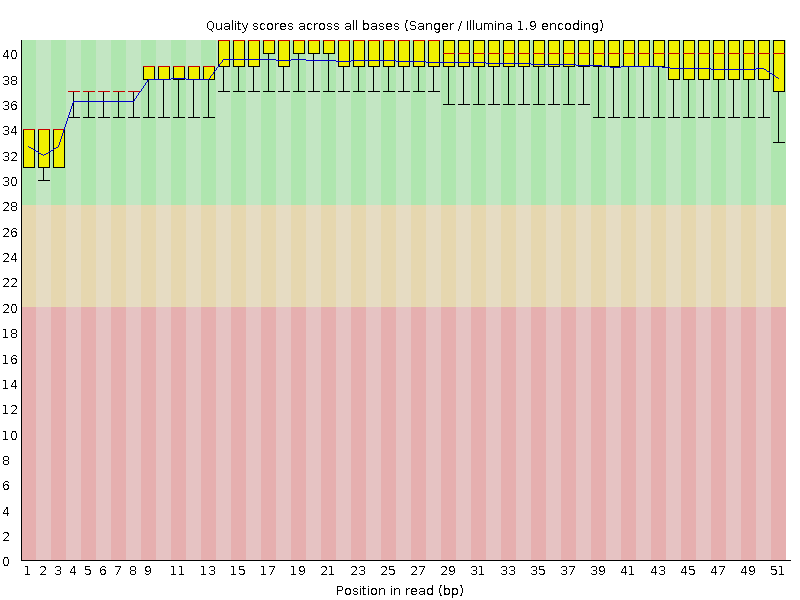
Per base quality после обработки trimmomatic.
Можно заметить, что trimmomatic не сильно повлиял на per base quality, число последовательностей уменьшилось с 17763 до 17612. Однако идеальным качество чтений транскриптома назвать нельзя. FastQC нашел проблемы с числом дубликатов последовательностей, а также с per base sequence content.
Часть II: картирование чтений
| Команда | Что делает | |
| В данной части все команды исполнялись аналогично практикуму 11. | ||
| hisat2-build chr8.fasta chr8 | Индексирует файл с референсной последовательностью. | |
| hisat2 --no-softclip -x chr8 -U chr8_good.fastq -S align.sam &> result.log | Выравнивает риды с референсной последовательностью, сохраняя информацию о работе программы в файл result.log. | |
| Параметр --no-spliced-alignment был удален, так как исследуется транскриптом, а в нём возможен различный сплайсинг. | ||
| samtools view align.sam -b -o align.bam | Преобразует .sam файл в бинарный .bam файл. | |
| samtools sort align.bam -T temp.txt -o align_sort.bam | Сортирует по возрастанию выравнивание чтений с референсом по координате в референсе в начале чтения. | |
| samtools index align_sort.bam | Индексирует отсортированное выравнивание. | |
Из файла result.log можно узнать следующие данные о картировании. 17307 ридов были картированы на референс 1 раз, 301 рид не был картирован. 4 рида были картированы более одного раза.
Часть III: Подсчёт чтений
Чтобы разобраться с параметрами программы htseq-count использовалась следующая страница: bioweb.pasteur.fr.
Значения опций:
- -f или --format: формат файла (.bam или .sam) с выравниванием (по умолчанию sam)/
- -i или --idattr: какой использовать атрибут GFF в качестве feature ID (по умолчанию gene_id).
- -s или --stranded: yes - считать только по прямой цепи, no - считать без учета цепи, reverse - считать только по обратной полследовательности.
- -m или --mode: определяет, как считать различное наложение рида на гены в том случае, когда это неоднозначно. Есть 3 режима: union, intersection-strict и intersection-nonempty. Риду присваивается свойство в зависимости от режима работы. Свойство no_feature означает, что рид не попал ни на один ген, свойство ambiguous - рид попадает на несколько генов и не может однозначно быть соотнесён с конкретным геном.
Чтобы проще разобраться в схеме работы различных режимов mode в статье прилагается следующее изображение:

Для запуска htseq-count с различными параметрами использовался следующий скрипт do.sh. В скрипте использовались следующие команды:
| Команда | Что делает |
| htseq-count -f bam align.bam -m |
Посчитывает, сколько раз на ген попали последовательности из ридов с различными параметрами. |
| | grep -w -v 0 > m_<mode>_s_<yes|no|reverse>_i_gene_id.count | Получает из STDOUT набор строк в формате |
По результатам вывода была составлена сводная таблица summary.xlsx.
| Feature | union + yes | union + no | union + reverse | strict + yes | strict + no | strict + reverse | nonempty + yes | nonempty + no | nonempty + reverse |
| ENSG00000104738.12 | 2 | 281 | 280 | 1 | 260 | 259 | 2 | 281 | 280 |
| ENSG00000253729.3 | 2 | 15816 | 15815 | 2 | 15388 | 15386 | 2 | 15816 | 15815 |
| __no_feature | 17303 | 1209 | 1212 | 17304 | 1659 | 1662 | 17303 | 1210 | 1212 |
| __not_aligned | 301 | 301 | 301 | 301 | 301 | 301 | 301 | 301 | 301 |
| __alignment_not_unique | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| _ambiguous | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Больше всего ридов попало на гены в режиме работы intersection-nonempty без учета направления цепи. Меньше всего генов было насчитано в режиме работы intersection-strict только на прямой цепи. Если воспользоваться схемой, расположенной выше, и данными из таблицы, можно прийти к выводы, что часть ридов частично выходила за пределы генов или находилось на месте разрыва генов (так как intersection-nonempty их посчитал, а intersection-strict - нет). 1 рид мог частично попасть на место наложения 2 генов, поэтому был отнесен к ambiguous в режиме union и к определенному гену в других режимах.
Обзор генов:
- ENSG00000104738.12: Minichromosome maintenance complex component 4 (MCM4) - 4ый компонент комплекса поддержки минихромосом - ДНК-хеликазы, необходимой для реплицакии ДНК.
- ENSG00000253729.3: Protein kinase, DNA-activated, catalytic subunit (PRKDC) - каталитический домен ДНК-активированной киназы. Этот белок необходим для репарации ДНК, в частности для устранения двуцепочечных разрывов.