Практикум 6. Чтение последовательностей по Сэнгеру
Ход работы
Файлы с хроматограммами для прямого и обратного прочтений в формате .ab1 с навзаниями L36_COI_F_F03_WSBS-Seq-07-10-16.ab1 и L36_COI_R_F04_WSBS-Seq-07-10-16.ab1 соответственно были взяты из папки P:\y17\term3\block2\ab1_files и переименованы в direct.ab1 и return.ab1 для удобства работы. Файлы доступны для скачивания по ссылкам. Полученные файлы были открыты в программе Chromas (Lite). Масштаб был настроен таким образом, чтобы обратная цепь находилсь строго под прямой. Обратное прочтение было инвертировано опцией Reverse. Затем были удалены нечитаемые проблемные участки с концов каждой хроматограммы в режиме Continuous edit (координаты в отчёте). Хроматограммы были просмотрены на предмет ошибок. Исправления вносились строчными буквами. Отредактированные хроматограммы были сохранены в формате .ab1, последовательности - в формате .fasta. Последовательности были выровнены командой:
needle direct_edit.fasta return_edit.fasta alignment.fasta -aformat3 fasta -auto
Затем полученное выравнивание было открыто в программе JalView и раскрашено по нуклеотидам. Наконец, консенсусная последовательность была получена командой:
consambig alignment.fasta result.fasta
Отчёт
- Исходные .ab1 файлы: direct.ab1 и return.ab1.
- Характеристики хроматограмм в целом:
- Прямое прочтение: длинный нечитаемый фрагмент вначале, более короткий в конце. В середине последовательности пики довольно четкие, спорных моментов мало. К концу хроматограммы высота шума увеличивается.
- Обратное прочтение: длинный нечитаемый фрагмент вначале, более короткий в конце. В середине последовательности сигналы довольно четкие по сравнению с шумом, спорных моментов мало. Пики в среднем ниже, чем при прямом прочтении.
- Координаты удалённых нечитаемых участков (относительно прямой цепи после ручного выравнивания):
- Прямое прочтение: 1 — 138; 672 — 713.
- Обратное прочтение: -34 — 3; 472 — 673.
- Отредактированные хроматограммы: direct_edit.ab1 и return_edit.ab1.
- Полученные последовательности: direct_edit.fasta и return_edit.fasta.
- Выравнивание последовательностей: alignment.fasta.
- Консенсусная последовательность: result.fasta.
- JalView проект: alignment.jvp.
- Таблица с описанием спорных моментов:
| Прочтение | № | Основание | Описание | Решение | Изображение |
| Прямое | 1 | 33 | Наложение нескольких сигналов одной высоты. | По обратной последовательности четко заметен аденин. | 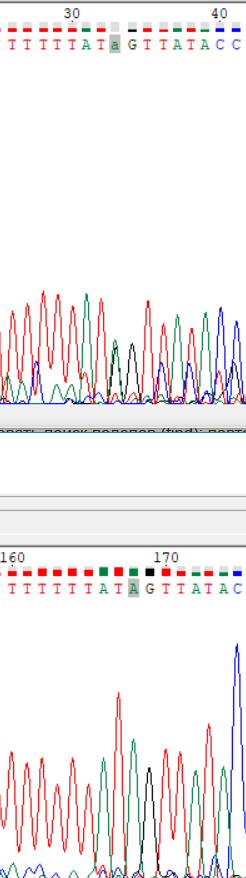
|
| Прямое | 2 | 81 | Высокий черный пик (гуанин), программой записан тимин. | На обратной последовательности указан тимин, исправление не требуется. | 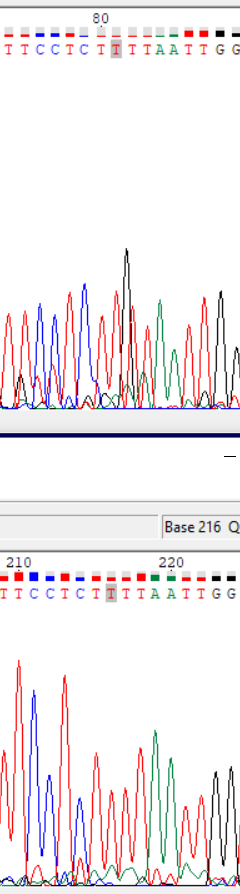
|
| Прямое | 3 | 163 | На хроматограмме заметен довольно четкий пик аденина, программа нуклеотид не распознала. | Обратное прочтение подтверждает наличие аденина. Исправить | 
|
| Прямое | 4 | 288 | Пик тимина оказался не сильно выше аденина, программа не смогла определить основание. | Обратное прочтение подтверждает наличие тимина. Исправление | 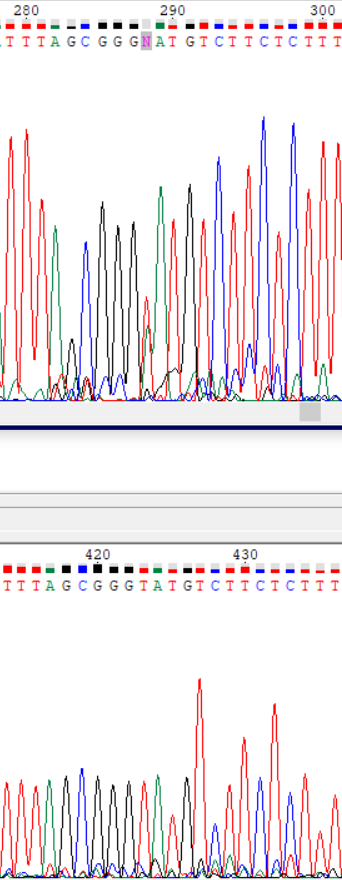
|
| Прямое | 5 | 73 | 2 равных по вышине пика гуанина и цитозина на небольшом расстоянии друг от друга. Программой не определены. | Обратное прочтение показывает, что на этом месте должен быть гуанин. Исправление. | 
|
Комментарий: координаты спорных моментов на обратном прочтении указаны следующим образом: <номер нуклеотида относительно прямого прочтения> (<номер нуклеотида на обратном прочтении>). Координаты на прямом протчении получаются вычитанием 135 из координат обратного прочтения.
| Прочтение | № | Основание | Описание | Решение | Изображение |
| Обратное | 1 | -19 (116) | Программа не определила нуклеотид по пику хроматограммы. Прямого прочтения для данного участка нет. | На хроматограмме четко заметен пик аденина. Исправление. | 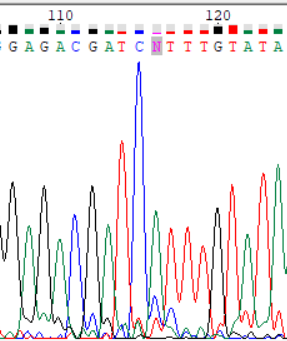
|
| Обратное | 2, 3 | 326 (461), 329 (464) | В первом случае почти полное наложение синего и зеленого пиков. Синий (цитозин) немного выше. Во втором случае красный пик заметно выше остальных, но программой не определен. | С помощью прямого прочтения подтверждается наличие цитозина и тимина в каждом случае соответственно. | 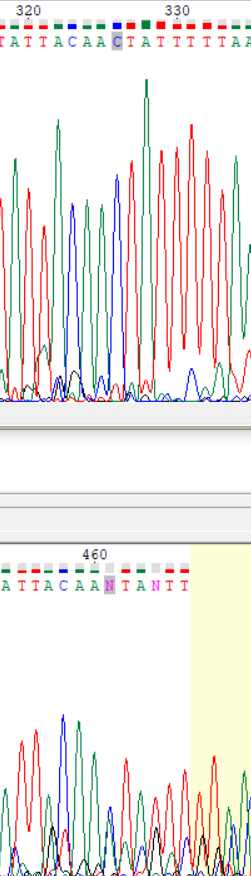 |
Нечитаемая хроматограмма
Поскольку использованная в задании хроматограмма оказалась довольно высокого качества, пришлось воспользоваться хроматограммой NN_G10.ab1, расположенной в папке bad. Изображение её фрагмента:
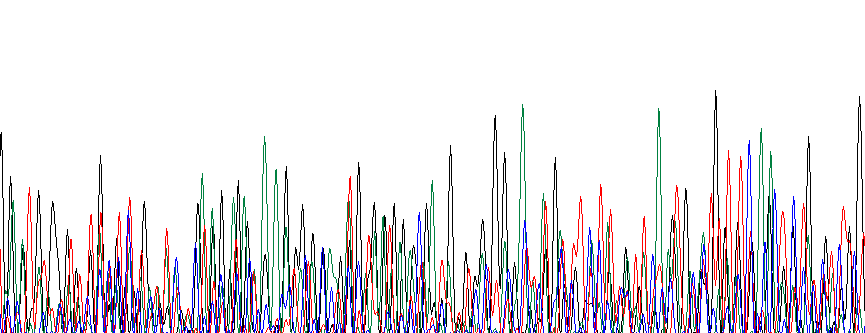
Вполне возможно, что причиной такой хроматограммы стала амплификация разных фрагментов ДНК, которые секвенировались одновременно. В результате на хроматограмме невозможно что-либо разобрать.