Занятие 9.
Цель данного занятия ознакомится с возможностями докинга низкомолекулярного лиганда в структуру белка В этом занятии мы будем пользоваться пакетом Autodock Vina и Autodock tools. Это программное обеспечение распространяется бесплатно для академических пользователей. Вы будете работать с белком лизоцимом структуру которого вы построили на основе гомологичного моделирования на прошлом практикуме. Программе Autodock Vina для докинга необходимы специально форматированные файлы pdb c зарядами и указанием торсионных углов. Для начала попробуем провести докинг одного из мономеров сахара (NAG) из прошлого занятия.
1.В банке pdb найдите SMILES нотацию для NAG.
CC(=O)N[C@@H]1[C@H]([C@@H]([C@H](O[C@H]1O)CO)O)O
2.C помощью obgen построим 3D структуру этого сахара в pdb формате. nag.pdb
3.Скриптом prepare_ligand4.py из пакета Autodock tools создайте pdbqt файл вашего лиганда. Так же, скриптом prepare_receptor4.py из пакета Autodock tools создайте pdbqt файл вашего белка.
4. Выберите атом сахара, который по Вашему мнению находится в центре сайта связывания и из текста pdb файла извлеките его координаты. Постройте файл vina.cfg с примерно таким содержанием:
center_x=41.401 center_y=42.525 center_z=27.262 size_x = 25 size_y = 25 size_z = 25 num_modes = 20
5.Теперь можно провести первый докинг:
vina --config vina.cfg --receptor prot.pdbqt --ligand nag.pdbqt --out nag_prot.pdbqt --log nag_prot.log
6.Просмотрим файл nag_prot.log и занесем в отчёт энергии 3ёх лучших расположений и геометрическую разницу между ними.
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.8 0.000 0.000
2 -5.6 2.648 5.646
3 -5.5 1.865 2.984
Все состояния на одной картинке (все достаточно компактно, ни одно из состояний не выбивается)
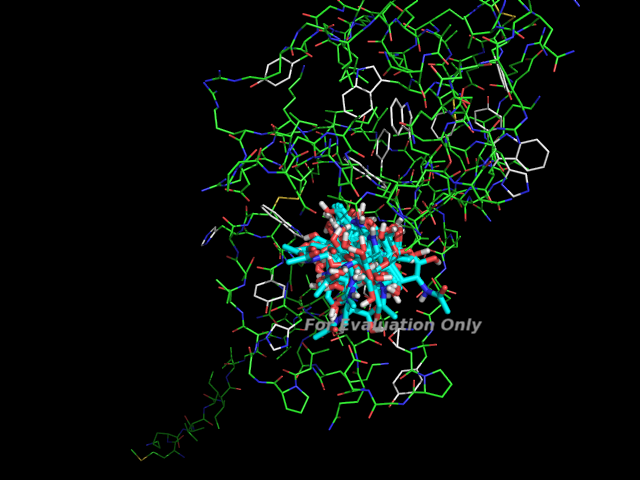
7.Теперь давайте проведём докинг рассматривая подвижность некоторых боковых радикалов белка. Сначала разобьем белок на две части, подвижную и неподвижную. Для подвижной части выберем 3 аминокислоты которые вы использовали в прошлом задании для позиционирования лиганда.
prepare_flexreceptor4.py -r prot.pdbqt -s GLU1_ASN5_ASP13и проведём докинг:
vina --config vina.cfg --receptor prot_rigid.pdbqt --flex prot_flex.pdbqt --ligand nag.pdbqt --out vina_prot_flex.pdbqt --log vina_prot_flex.log
8.Просмотрим файл nag_prot_flex.log и занесем в отчёт энергии 3ёх лучших расположений и геометрическую разницу между ними.
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -4.8 0.000 0.000
2 -4.7 1.328 2.219
3 -4.4 1.239 3.568
Все состояния на одной картинке (несколько состояний выбиваются из общего расположения,возможно, надо было
уменьшить размер ячейки места связывания)
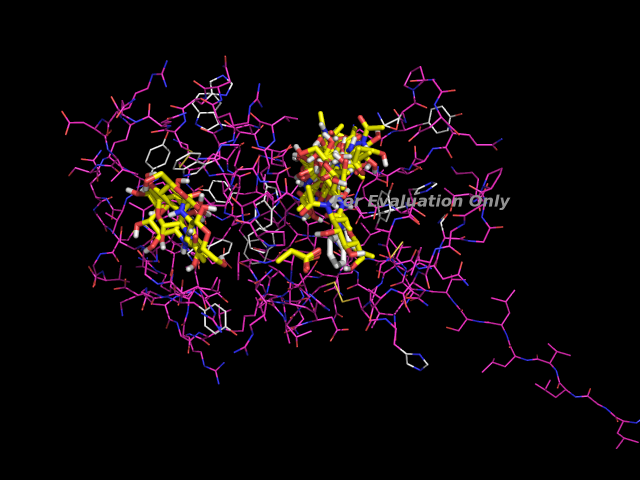
9. Докинг не смог расположить лиганд, как в модели. Если смотреть в PyMol, то ни одно из состояний, полученных докингом не совпало с моделью. Но лучше получился простой докинг (по минимуму энергии -5.8), чем разделение на подвижную и неподвижную части.
10.NAG содержит в себе СH3C(=O)NH группу. Создайте 3 лиганда где метильный радикал этой группы будет заменён на OH, NH2,H. Для каждого из этих лигандов проведите обыкновенный докинг и представьте результаты в виде таблицы из трёх лучших расположений для каждого лиганда.
1)
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.7 0.000 0.000
2 -5.7 2.689 5.728
3 -5.6 2.043 4.734
2)
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.7 0.000 0.000
2 -5.6 2.376 5.024
3 -5.5 1.773 3.113
3)
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.3 0.000 0.000
2 -5.2 1.861 3.246
3 -5.1 2.066 3.710
По значению энергии видим, что замена на другую группу не существенно меняет связывание, кроме замены на водород.
У него энергия больше всех, т.к. он не обеспечивает взаимодействия с лиганда с белком.