Занятие 7-8.
Цель данного занятия ознакомится с возможностями моделирования молекулярной динамики. В этом занятии мы будем пользоваться пакетом молекулярной динамики Gromacs. Это программное обеспечение распространяется под лицензией GPL, т.е. пользователь может скачать исходный код и свободен его изменять по своему усмотрению.
Типы файлов:
gro - файл с координатами системы.
top - файл с описанием ковалентных и нековалентных взаимодействий в молекулах.
mdp - файл с описанием параметров для работы молекулярно-механического движка.
tpr - файл для молекулярно-механического движка по сути есть объединение gro, top и mdp.
trr, xtc - файл с координатами после расчёта.
Объекты для практикума
Моделирование перехода ДНК из А в В форму
Вам даны файлы:
файл праметров для минимизации энергии em.mdp.
файл праметров для "утряски" воды pr.mdp pr.mdp.
файл праметров для молекулярной динамики md.mdp.
1. Зайдем на kodomo через Putty и перейдем в рабочую директорию.
cd Starovoytova/md
2. С помощью программы fiber из пакета 3DNA построим небольшой дуплекс с последовательностью GATCTA. Для того, что бы fiber работал надо задать путь и переменную среды:
export X3DNA=/home/preps/golovin/progs/X3DNA
export PATH=/home/preps/golovin/progs/X3DNA/bin:${PATH}
3. Скопируем директорию Starovoytova на 172.16.0.140. Зайдем по ssh или Putty на 172.16.0.140. Теперь построим файл топологии системы в силовом поле amber99sb и файл с координатами в формате Gromacs. Предполагается, что структура дуплекса находится в файле dna.pdb.
pdb2gmx -f dna.pdb -o dna -p dna -ff amber99sb -water tip3pЕсли вы получили сообщение об ошибке, то удалите 5' фосфаты из структуры дуплекса. Их два на 5' конце кажой цепи. Вам также надо изменить имена нуклеотидов добвавив D к названию нуклеотида," Т"->"DT" (пример для vim: :%s/ \([GATC]\) \([AB]\)/ D\1 \2/). И наконец замените имя атома "С5М" на " С7".
4. Сделаем небольшой отступ в ячейке от ДНК.
editconf -f dna.gro -o dna_ec -d 1.55. Проведём оптимизацию геометрии системы, что бы удалить "плохие" контакты в молекуле.grompp -f em -c dna_ec -p dna -o dna_em -maxwarn 1 mdrun -deffnm dna_em -v6. Добавим в ячейку молекулы воды.genbox -cp dna_em -p dna -cs -o dna_s7. Нейтрализуем заряд системы. Это делаем в два шага: строим tpr и запускаем genion.grompp -f em -p dna -c dna_s -o dna_s genion -s dna_s -o dna_si -p dna -np Xгде Х это количество положительных ионов необходимых для нейтрализации заряда системы. 8. Проведём "утряску" воды:grompp -f pr -c dna_si -p dna -o dna_pr -maxwarn 1 mdrun -deffnm dna_pr -v9. Переформатируем dna_pr.gro и dna_si.gro в pdb формат.
Видим, что на первой картинке молекулы воды расположены упорядоченно. После утряски воды они расположены хаотично, сама цепь ДНК своего положения не потеряла. 10. Теперь надо скопировать ваши файл на суперкомпьтер. Сначала зайдем на суперкомпьтер и создадим папку с Вашей фамилией и вернёмся на kodomo. ssh skif mkdir Starovoytova exit Копируем файлы: cd .. scp -r md/* skif:Starovoytova/ 11. Запускаем тестовое моделирование на суперкомпьтере. ssh skif cd Starovoytova grompp -f md -c dna_pr -p dna -o dna_md -maxwarn 1 mpirun -np 16 -q test -maxtime 5 /home/golovin/progs/bin/mdrun_mpi -deffnm dna_md -v Просмотреть ход счёта можно в файле mdrun_mpi.out-.... less mdrun_mpi.out-.... Нажмите shift+. для перехода в конец файла. Если файл не содержит ошибок, то переходим дальше: 12. Запускаем основное моделирование на суперкомпьтере. mpirun -np 16 -maxtime 1200 /home/golovin/progs/bin/mdrun_mpi -deffnm dna_md -v
ПараметрыСиловое поле используемое при построении топологии - amber99sb Заряд системы. Причины - 0. Нейтрализуем отрицательный заряд ДНК при добавлении воды. Размер и форма ячейки - кубическая, 5.11800 х 4.94600 х 5.49400 Минимизация энергии - - Алогритм минимизации энергии - integrator = l-bfgs - Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий - двойное обрезание (cut-off) Модель, которой описывался растворитель - implicit_solvent = No Утряска растворителя (pr.mdp)- - Для биополимеров, укажите параметр который обуславливает неподвижность биополимера - define = -DPOSRES - Число шагов - nsteps = 10000 - Длина шага - dt = 0.001 ; в pс - Алгоритм расчёта электростатики- pme и Ван-дер-Ваальсовых взаимодействий - cut-off - Алгоритмы термостата- Berendsen и баростата - нет. - Основной расчёт МД (md.out) -Время моделирования, количество процессоров, эффективность маштабирования. (mdrun_mpi.rep-240950)4 hours 39 minutes 18 seconds, 16, 100% (mdrun_mpi.rep-240950) -Длину траектории (=число шагов*длину шага) = 10000 пс. -Число шагов = 5000000. -Длина шага = 0,002 пс. -Алгоритм интегратора - мд. -Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий. - pme (суммирование по Эвальду) - двойное обрезание (cut-off). -Алгоритмы термостата и баростата. Для температуры и давления - метод Berendsen.
Анализ результатов 1Любой анализ начинают с визуального анализа движений молекул. При вопросе о выводе групп выберите DNA.
trjconv -f dna_md.xtc -s dna_md.tpr -o dna_pbc_1.pdb -skip 20 -pbc molОткроем b_pbc_1.pdb в PyMol. Если не устроил результат визуализации попробуйте:trjconv -f dna_md.xtc -s dna_md.tpr -o dna_fit_1.pdb -skip 20 -fit rot+transТаким образом было получено два файла dna_pbc_1.pdb и dna_fit_1.pdb. Более удобным для анализа является файл dna_fit_1.pdb, т.к. молекула находится в центре и не прыгает по углам. На 800 пикосекунде происходит образование В-формы.
2Определим средне-квадратичное отколнение в ходе моделирования. Так как у нас происходит конформационный переход сначала расчитаем отклонение в ходе все симуляции относительно стартовой структуры.
g_rms -f dna_md.xtc -s dna_md.tpr -o rms_1И относительно каждой предыдущей структуры на растоянии 400 кадров. Если ближе к концу закончился конформационный переход, то отколнение должно уменьшаться.
g_rms -f dna_md.xtc -s dna_md.tpr -o rms_2 -prev 400Наиболее сильное отклонение происходит относительно предыдущих структур. 3Определим изменение гидрофобной и гидрофильной поверхности в ходе конформационного перехода.
g_sas -f dna_md.xtc -s dna_md.tpr -o sas_dna.xvgПостроим зависимость изменения гидрофобной (красным) и гидрофильной (зеленым) поверхностей доступных растворителю от времени.площади гидр. и гидроф. поверхностей почти не меняются при переходе. Конформационный переход из А в В форму происходит, т.к. А-форма менее устойчива, чем В-форма 4Количество водородных связей:(красным 1:2 столбики, 1:3 зеленым)
ДНК-ДНК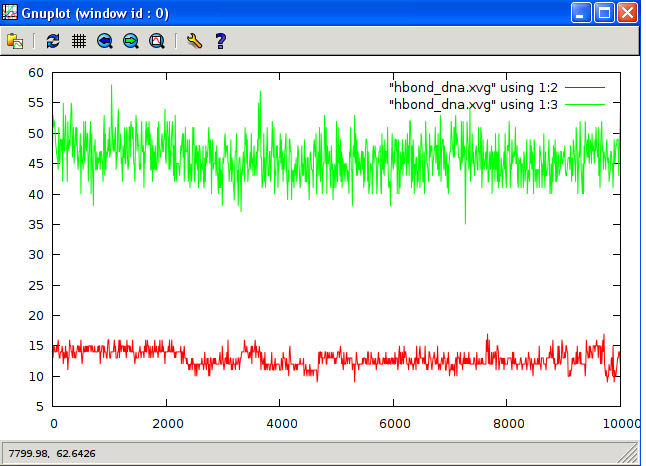
ДНК-вода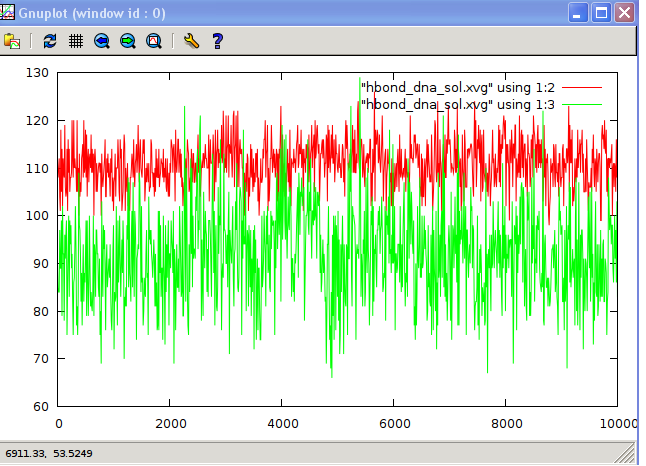
Красным- водородные связи. ДНК-ДНК: связей должно быть 14. Видим, что колебания как раз в районе 14. С растворителем число водородных связей колеблется от 100 до 120. Т.е меняется не сильно, что не мешает переходу из А в В форму.