Докинг низкомолекулярных лигандов в структуру белка
- Подготовка файлов
В банке pdb найдем SMILES нотацию для NAG, эту нотацию в файл nag.smi. C помощью obgen построим 3D структуру этого сахара в pdb формате.
nag.pdb
Скриптом prepare_ligand4.py из пакета Autodock tools создадим pdbqt файл лиганда nag.pdbqt. Срипт расположен в директории, укажем путь: export PATH=${PATH}:/home/preps/golovin/progs/bin. Так же, скриптом prepare_receptor4.py из пакета Autodock tools создадим pdbqt файл белка LYS_BOMMO 1.pdbqt. - Докинг
Итак, есть входные файлы. Теперь надо создать файл с параметрами докинга vina.cfg. Удобно область структуры белка, в которой будет происходить поиск места для связывания, задать как куб с неким центором. Координаты центра определим из модели комплекса, которую построили на прошлом занятии. Определяется центр масс с помощью PyMol, команда pseudoatom.
Построим файл vina.cfg
Докинг:
vina --config vina.cfg --receptor 1.pdbqt --ligand nag.pdbqt --out nag_prot.pdbqt --log nag_prot.logТри лучшие расположения (энергии 3ёх лучших расположений и геометрическая разница между ними)
mode | affinity | dist from best mode | (kcal/mol) | rmsd l.b. -----+------------+---------- 1 -4.9 0.000 2 -4.7 2.243 3 -4.6 1.426В PyMol загрузим файлы nag_prot.pdbqt и 1.pdbqt. Включим анимацию. Все состояния отображены на картинке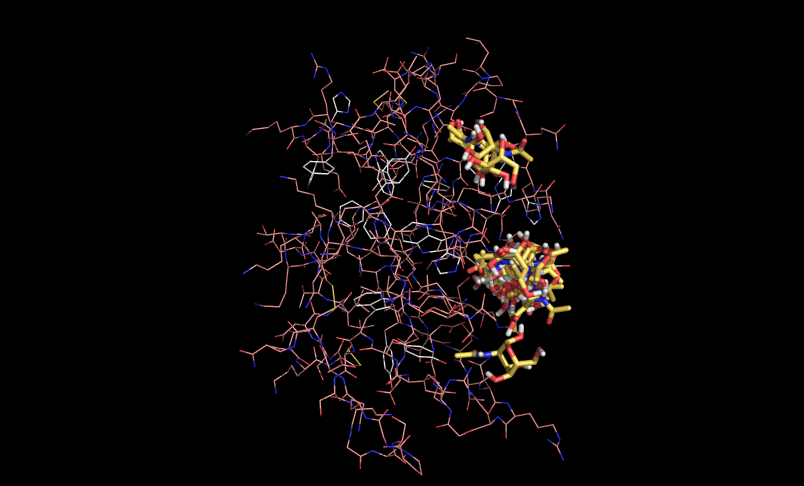
- Проведём докинг, рассматривая подвижность некоторых боковых радикалов белка.
Сначала разобьем белок на две части, подвижную и неподвижную. Для подвижной части выберем 3 аминокислоты, которые использовали в прошлом задании для позиционирования лиганда.
python /usr/share/pyshared/AutoDockTools/Utilities24/prepare_flexreceptor4.py -r 1.pdbqt -s ALA101_ASN43_TRP60
Аланин программа не захотела брать, поэтому для подвижной части - только 2 аминокислоты.
vina --config vina.cfg --receptor 1_rigid.pdbqt --flex 1_flex.pdbqt --ligand nag.pdbqt --out vina_1_flex.pdbqt --log vina_1_flex.logТри лучшие расположения (энергии 3ёх лучших расположений и геометрическая разница между ними)
mode | affinity | dist from best mode | (kcal/mol) | rmsd l.b. -----+------------+---------- 1 -4.9 0.000 2 -4.8 1.797 3 -4.7 1.781В PyMol загрузим файлы vina_1_flex.pdbqt и 1_rigid.pdbqt. Включим анимацию. Все состояния отображены на картинке. Над лигандом в белке выделены ASN (слева от лиганда, цвета magenta как и лиганд) и TRP (справа, белого цвета).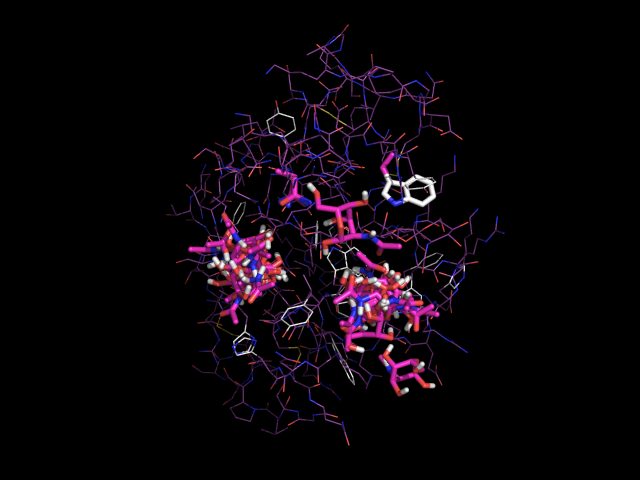
В принципе лиганд почти также перемещается на анимации, как и в первом случае. А еще можно увидеть, что в процессе докинга меняют свое положение и аминокислота ASN. Но у этой аминокислоты не 20 состояний, а ровно столько сколько состояний лиганда, когда он подходит близко к ASN.
- Докинг не близко располагает лиганд на то место, как получили в моделировании: доказательство на картинке ниже. Показано лучшее состояние 4 (бирюзовый) с энергией -4.6 kcal/mol, ASN43 (желтый), TRP60 (белый), лиганд при моделировании (magenta).
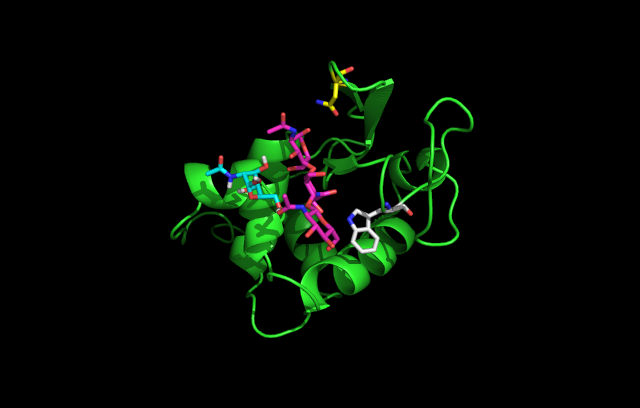
Моделирование структур биополимеров
© Migur Anzhela 2012