
Я рассматривала структуры 7CHQ и 7XIS. Первая структура разрешена методом РСА, а вторая определена с помощью ЯМР. Это структуры анти-CRISPR AcrIE2 - ингибитора системы CRISPR-Cas. Такие белки производятся фагами и другими мобильными генетическими элементами для защиты от CRISPR-Cas-опосредованную деструкциии в бактерии. [1] AcrIE2 ингибирует тип I-E CRISPR-Cas системы Синегнойной палочки (Pseudomonas aeruginosa).
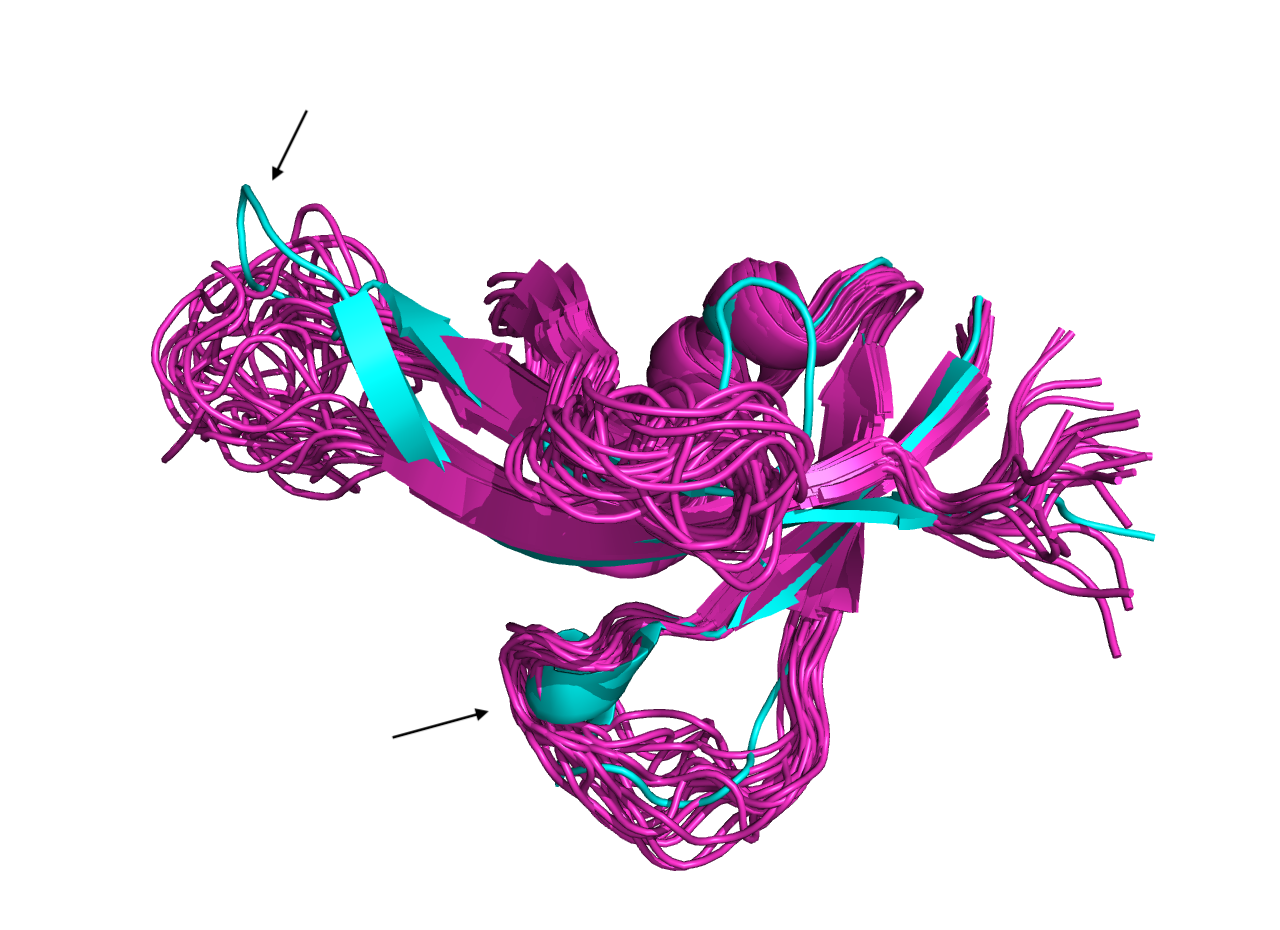
Для структур, разрешенных ЯМР, важным параметром является число моделей. Для 7XIS доступно 20 моделей (посчитано 50, из них отобраны 20 с более низкой энергией). Струкутра 7CHQ получена с высоким разрешением 1.33 ангстрем при полноте 99,6%. На Рисунке 1 представлены структуры этого белка, разрешенные разными методами и наложенные друг на друга.
В первую очередь бросилось в глаза отсутвие молекул воды в структуре ЯМР. Также в структуре ЯМР есть водороды, это ожидаемо. В целом струкутры похожи между собой, наблюдается хорошее совпадение паттернов консервативных структур (альфа-спиралей, бета-структур). На правом рисунке я привела пример зон, которые выбиваются при наложении структур. По-разному размечены начало и конец альфа-спиралей и бета-листов. Кроме того, по-разному идут некоторые белковые петли. Кстати, если посмотреть на результаты Validation, то оказывается, что для NMR-структуры характерен больший процент маргинальных остатков (по картам Рамачандрана и ротамерам).
Расхождения можно объяснить тем, что белки находились в разных условиях. ЯМР более нативный метод, данные конформации соответствуют белку в растворе. Построение множества моделей может отражать подвижность, но в общем случае скорее свидетельствует о неточности метода. При проведении РСА белок находится в составе кристалла. При этом кристалл получали при температуре 20 градусов (а не при криотемпературах) и такие структуры часто лучше соответствуют структурам, полученным ЯМР. [2]
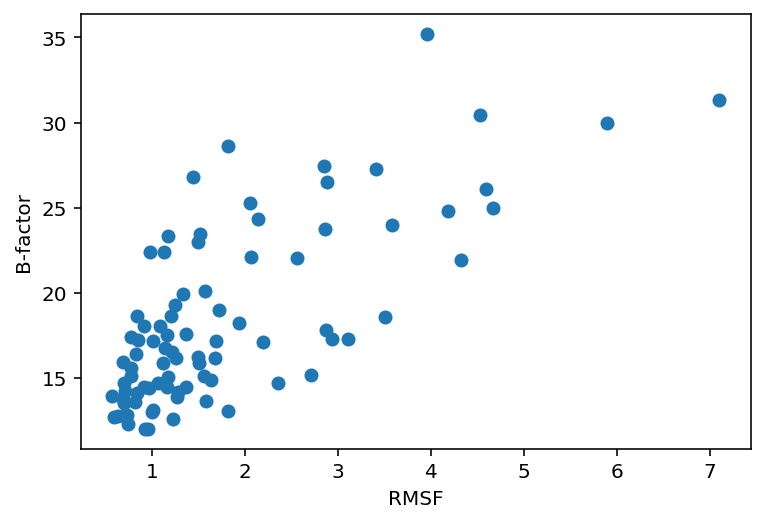
В этом задании мы представили, то различия в моделях обусловлены именно подвижностью, а не шумом и неполнотой данных. Мерой подвижности белка служит параметр RMSF (root-mean-square fluctuation) - это среднее отклонение частицы (или аминокислотного остатка) во времени по сравнению с референсной позицией (усредненная во времени позиция). Таким образом, с помощью RMSF можно анализировать части структуры, флуктуация которых от средней позиции по времени больше (или меньше). [3]
Сначала я построила выравнивание между последовательностью аминокислот, соответствующих двум структурам. Для ЯМР я оставила одну модель на этом этапе (которая выгружается при операции fetch 7XIS). Выравнивание было выполнено в Pymol с помощью инструмента A -> align и отображения последовательностей S. Интересно, что на данном этапе можно не только визуально оценить выравнивание и найти недостающие остатки, а также увидеть, какие остатки в пространстве расположены по-разному (по мнению сервера). На Рисунке 2 я привела пример участка выравнивания. Единственное различие в последовательностях - длина His-тэга, его я и так отсеку при выполнении задания. Таким образом, я взяла остатки с 1 по 86.

Затем я сравнила, как связаны значение B-фактора и параметр подвижности RMSF. Для структуры 7CHQ я для каждого остатка посчитала среднее значение B-фактора по каждому аминокислотному остатку. Для структуры 7XIS, полученной ЯМР, я посчитала RSMF для каждого соответствующего аминокислотного остатка. На Рисунке 3 изображен график зависимости этих двух величин. Можно заметить, что зависимость отклоняется от линейной и что B-фактор и RMSF положительно коррелируют: большему B-фактору, как правило, соответствует большее значение RMSF. Если считать B-фактор отражением подвижности, то можно сказать, что множество моделей ЯМР отражают динамику белка.
В этом задании я сравнивала наличие водородных связей в обеих структурах, разрешенных разными методами. Для этого я выбрала водородные связи, разные по расположению в белке и по взаимодействующим группировкам.
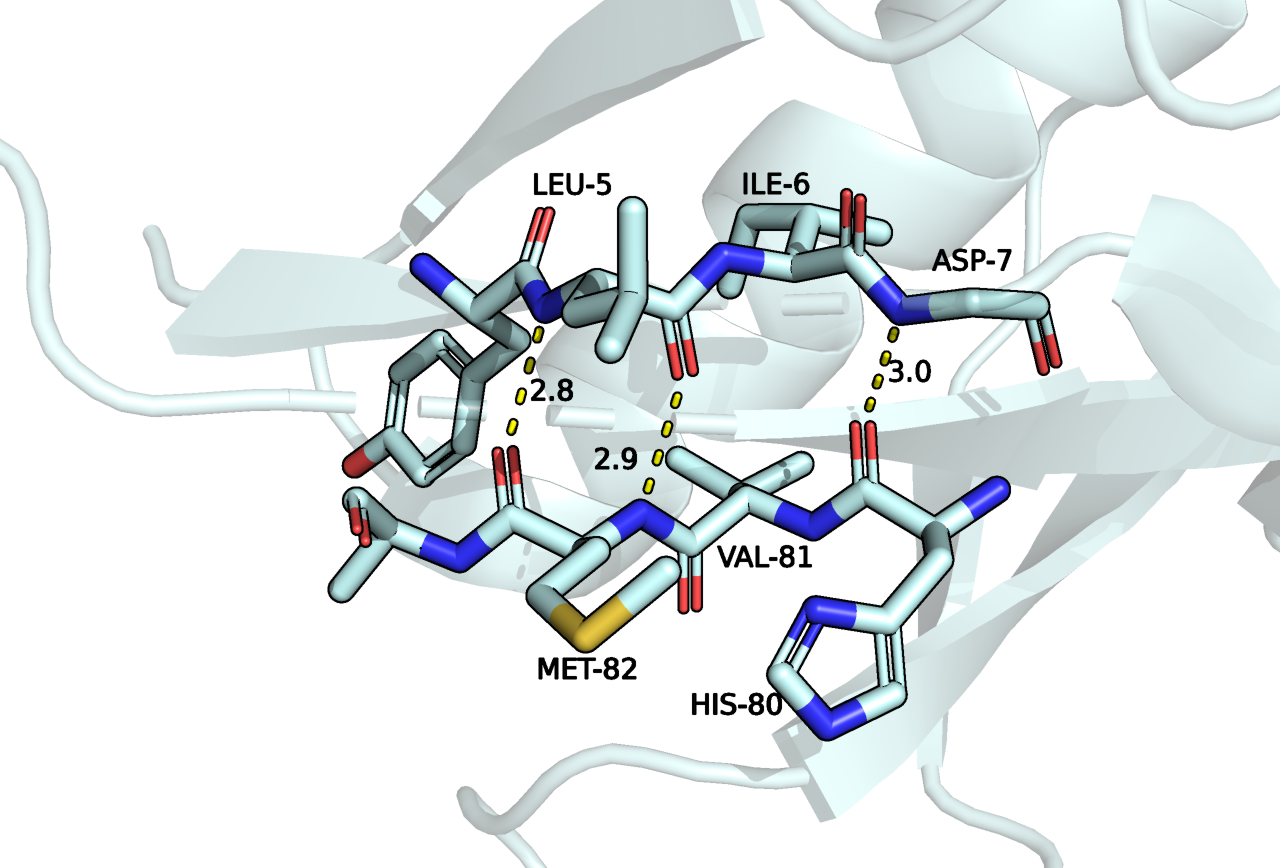
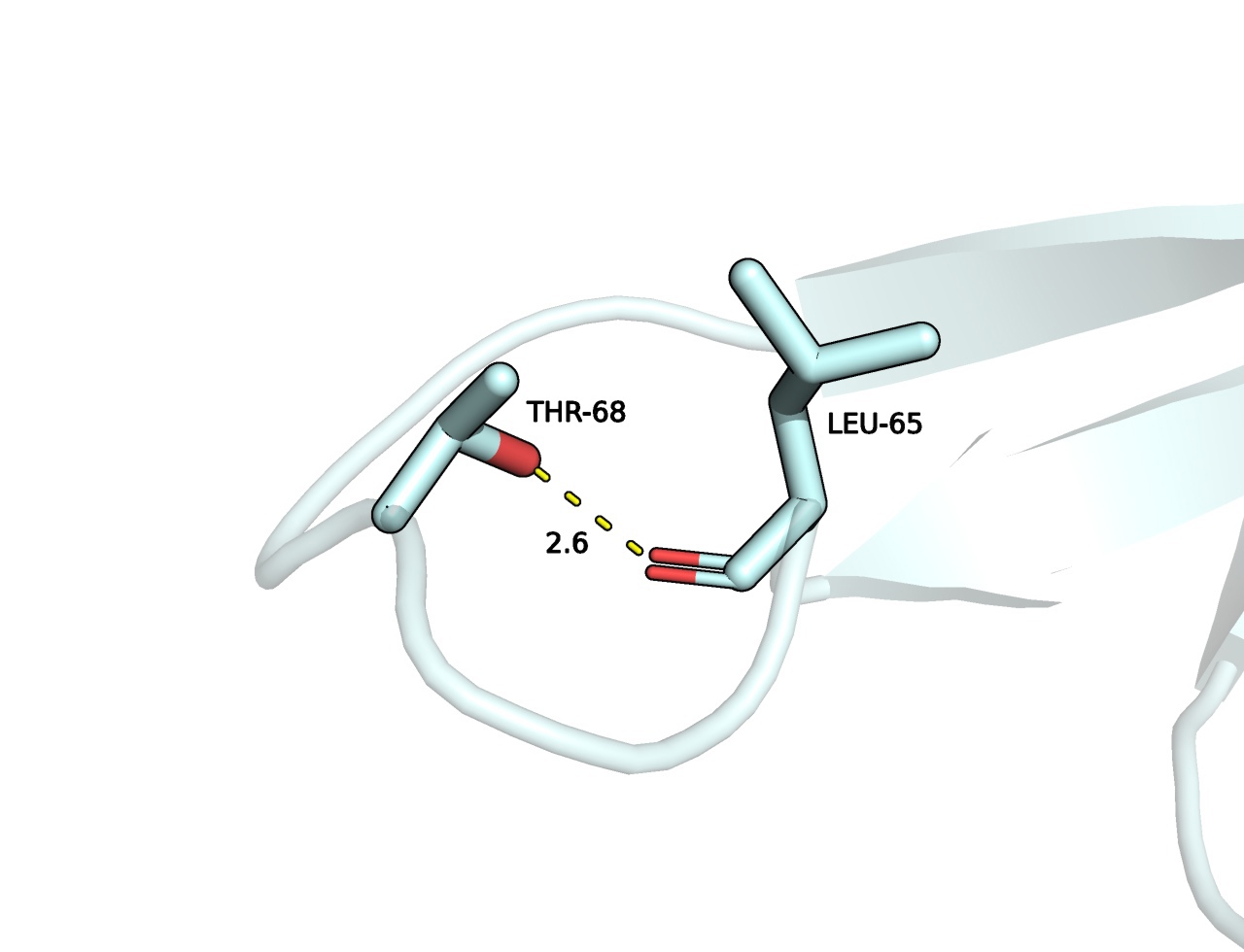
Для каждой пары атомов (донор и акцептор), между которыми в РСА наблюдается водородная связь, было определено расстояние для каждой модели, полученной ЯМР. Отсутствие или наличие водородной связи определялось только по расстоянию, предполагалась связь при дистанции < 3.5 ангстрем. Скорее всего водородных связей предсказалось больше, чем есть на самом деле, поскольку в нашей модели не накладывались ограничения на углы.
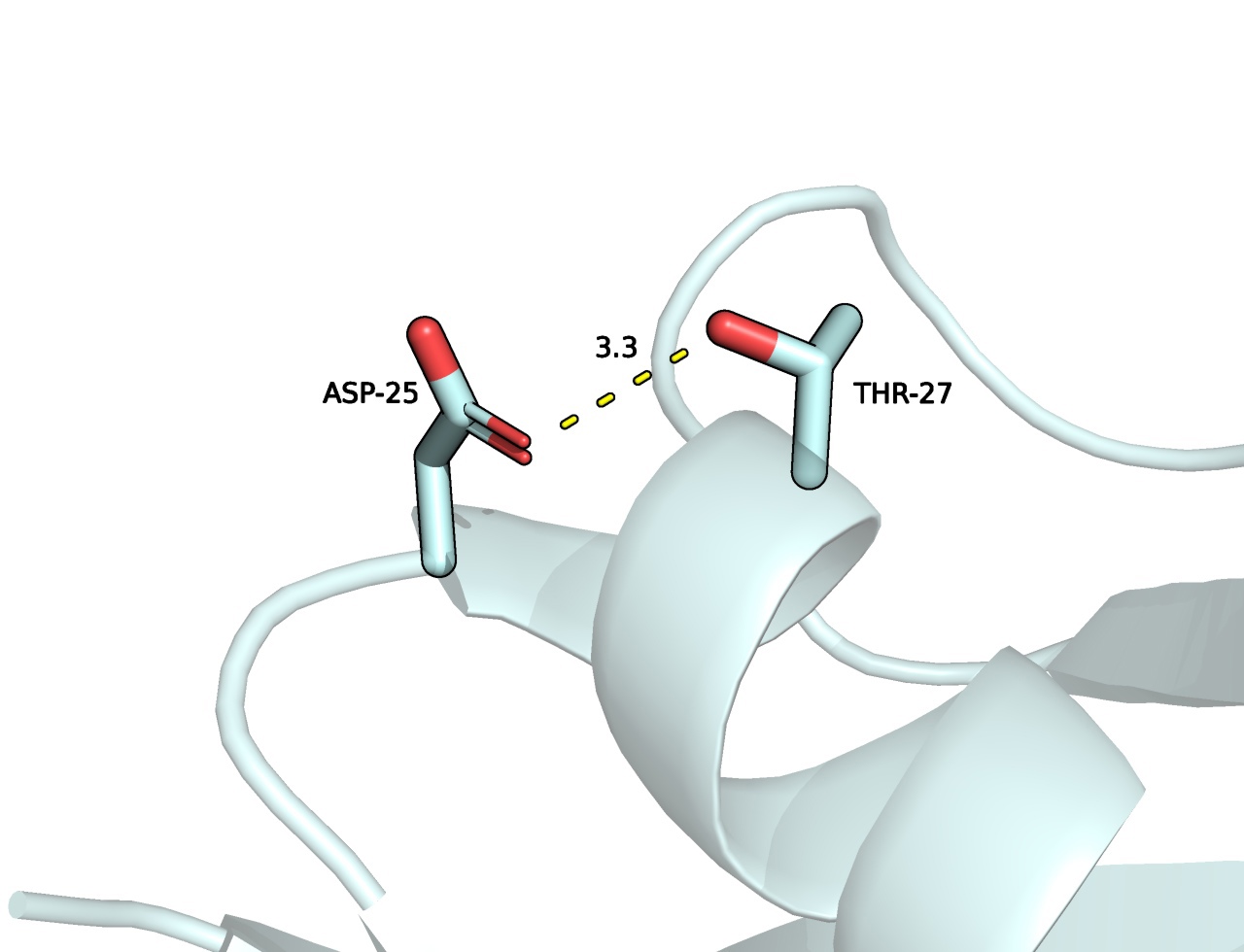
Для водородной связи между Asp-25 и Thr-27 проверяла наличие связи донора с обоими кислородами и брала наименьшую из них.
Получается, что информация о связей между атомами остова хорошо совпадает в моделях, построенных разными методами. Это консервативные и менее подвижные структуры. При анализе связей между аминокислотными остатками, принадлежащими петлям, водородные связи не обнаруживаются в ЯМР моделях.
| Связь | Расстояние в РСА | Минимальное расстояние в ЯМР | Максимальное расстояние в ЯМР | Медианное расстояние в ЯМР | Моделей со связью (из 20) | Моделей со связью, (%) |
| LEU5-MET82 | 2.942 | 2.870 | 3.315 | 3.136 | 20 | 100 |
| ASP25 -THR27 | 3.298 | 2.643 | 5.788 | 5.201 | 4 | 20 |
| LEU65-THR68 | 2.561 | 3.675 | 10.873 | 7.25 | 0 | 0 |
[1] Anti-CRISPR AcrIE2 Binds the Type I-E CRISPR-Cas Complex But Does Not Block DNA Binding
[2] A method for validating the accuracy of NMR protein structures