Потенциал-зависимый анионный канал, или митохондриальный порин (Voltage-dependent anion channel, VDAC) - это самый представленный белок внешней мембраны митохондрии эукариот. Этот анионный канал ведет себя как диффузионная пора для небольших гидрофильных молекул. Канал принимает открытую конформацию при низком или нулевом мембранном потенциале и закрытую конформацию при потенциалах выше 30-40 мВ по модулю. Хотя оба состояния допускают прохождение простых солей, VDAC в закрытом состоянии перестает пропускать органические анионы (АТФ, АДФ, пируват, малат и другие). Предположительно, изменение напряжения приводит к сдвигу фрагмента белка из канала и уменьшению эффективного радиуса поры. Кроме роли в регуляции метаболизма, VDAC является ключевым игроком митохондриально-индуцированного апоптоза. [0, 1]
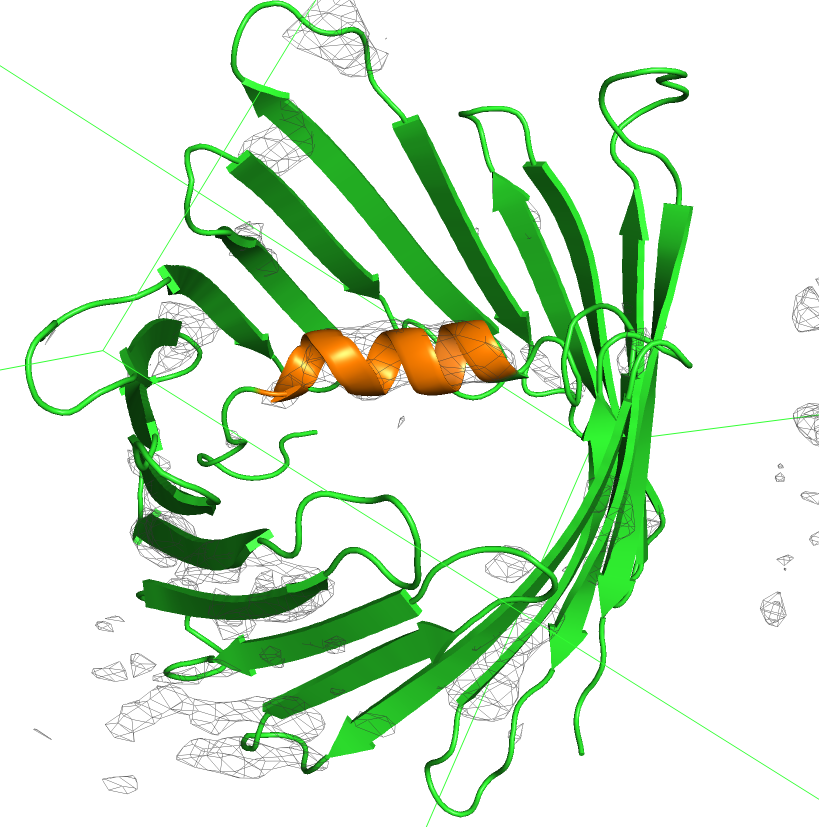
У человека, как и у большинства других высших эукариот, экспрессируются 3 различных VDACs: VDAC1, VDAC2 и VDAC3.В этом практикуме я рассматривала структуру митохондриального порина 1 человека (VDAC1). Фермент был открыт в 1976 году, и в 2008 году независимыми лабораториями были проведены три эксперимента по расшифровке структуры. Первая структура была разрешена методом мультимерного NMR, вторая - с помощью гибридного подхода с разрешением 4.1 Å (анализирую в этом практикуме, 2JK4). Третьей разрешили мышиную VDAC-1 с разрешением 2.3 Å. [4]. Наверняка каждые авторы стремились первыми рассказать о структуре (сейчас мы знаем, кто преуспел). Все эти статьи описательные: рассматривается структура фермента и его димеризация, обсуждается N-концевая спираль и её нестабильность. VDAC1 человека представляет из себя бета-бочонок, сложенный из 19 бета-листов с альфа-спиралью, лежащей горизонтально, примерно посередине поры. Его структура представлена на Рисунке 1.
Структура 2JK4 , которую я далее буду рассматривать, была получена с помощью X-ray. Однако в оригинальной статье отметили недостаточное качество полученной электронной плотности и для получения конечной структуры применяли «гибридный» подход: сочетали информацию полученную методом ЯМР-спектроскопии (NMR) и рентгеновской кристаллографии (X-ray). [1] То есть мы не ожидаем хорошего качества выложенных в PDB данных и учитываем, что только по ним структуру не восстанавливали. Cчитается, что эти сочетание NMR + X-ray улучшают качество полученной структуры и выгодно друг друга дополняют. Дифракция X-ray лучше отражает форму всей молекулы, а NMR - детали расположения атомов. [2]
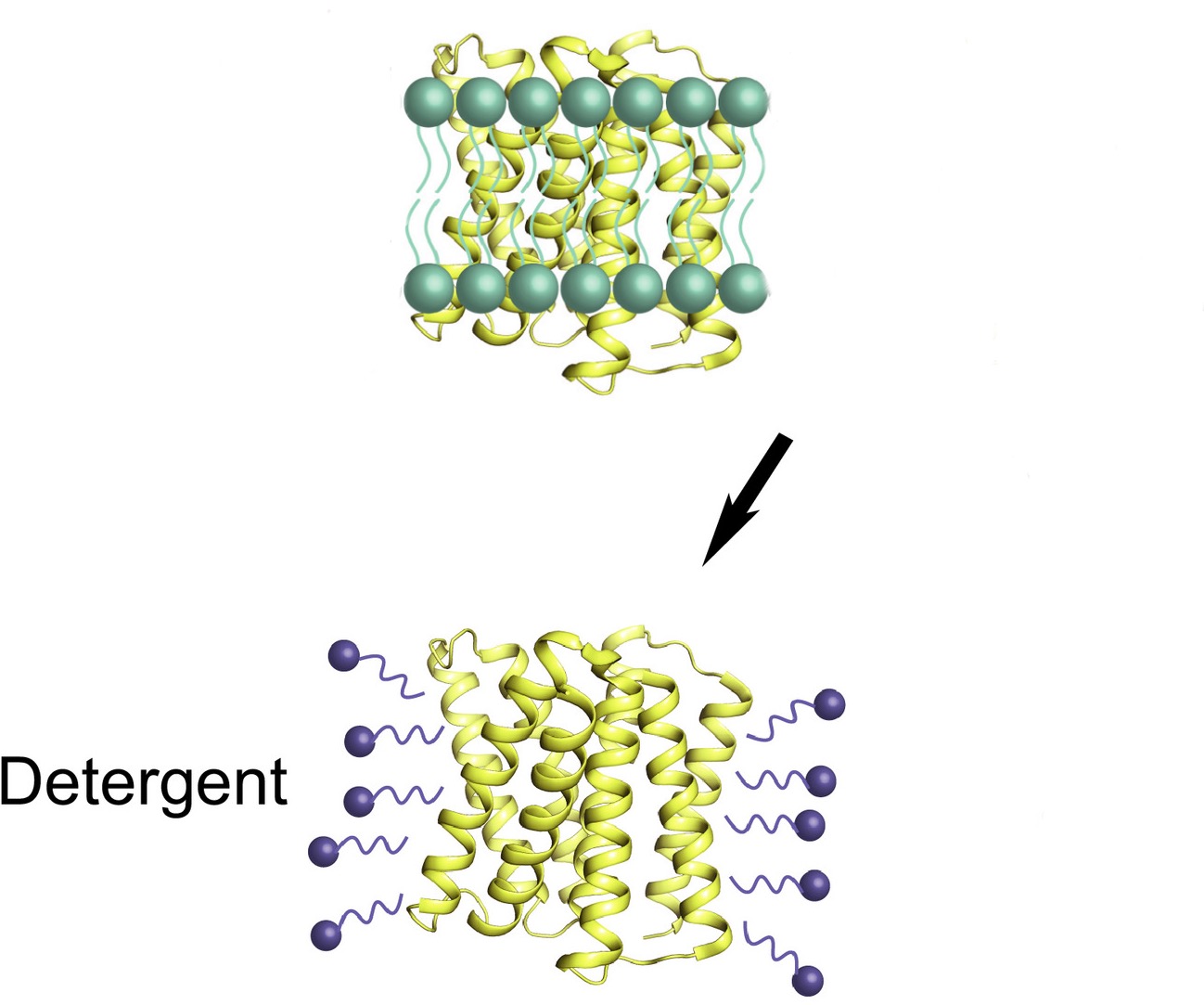
Трудности в расшифровке этого фермента заключается в том, что это мембранный белок. Для кристаллизации белка применялся скрининг детергентов (подобрали Cymal-5). Фермент в правильно подобранных условиях должен правильно свернуться заново. Кристаллы, таким образом, представляют из себя регулярную структуру из мембранных белков и молекул детергента. [3] ( Рисунок 2)
Оценка качества структуры
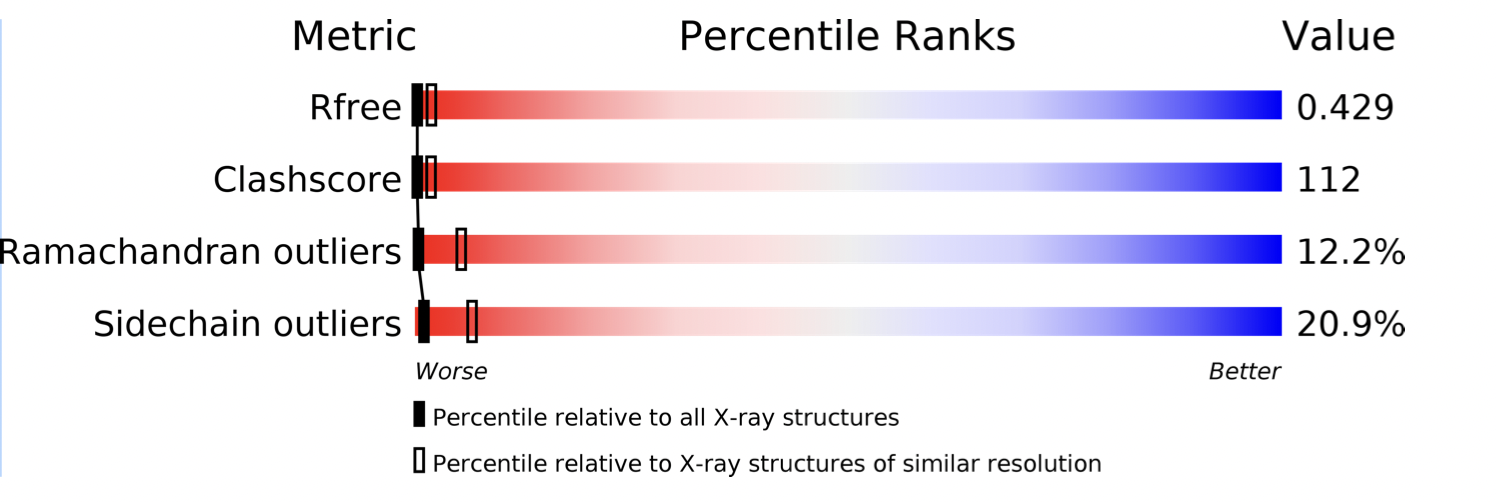
Сначала взглянем на глобальные валидационные матрики и сравним с качеством других структрур, хранящихся в PDB.
Начнем с того, что разрешение анализируемой структуры низкое (4.1 Å) при полноте данных 99.5%. Поэтому на фоне всех структур она всегда будет смотреться плохо (Рисунок 3). Однако, даже среди структур с близким разрешением (это диапазон ~ от 4.5 до 3.7 Å) эта структура отличается крайне низким качеством.
Качество не очень хорошее, поэтому затруднительно восстановление структуры. Можно заметить, что более плотные участки (mesh = 3) определились для внутренней альфа-спирали, у нее мы ожидаем относительно более выское качество (Рисунок 1). Во многих случаях положения остатков не подтверждены ЭП, пример такой ситуации преставлен на Рисунке 4.
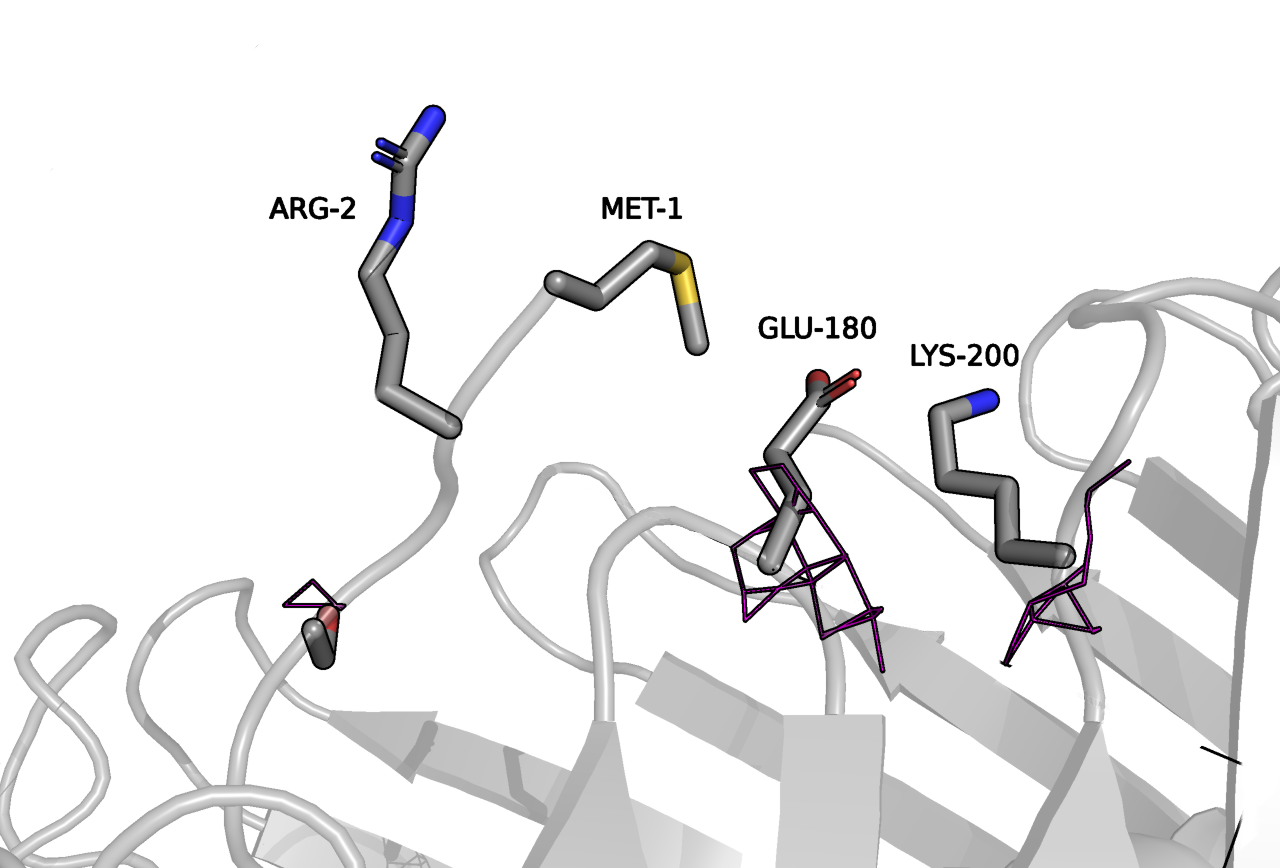
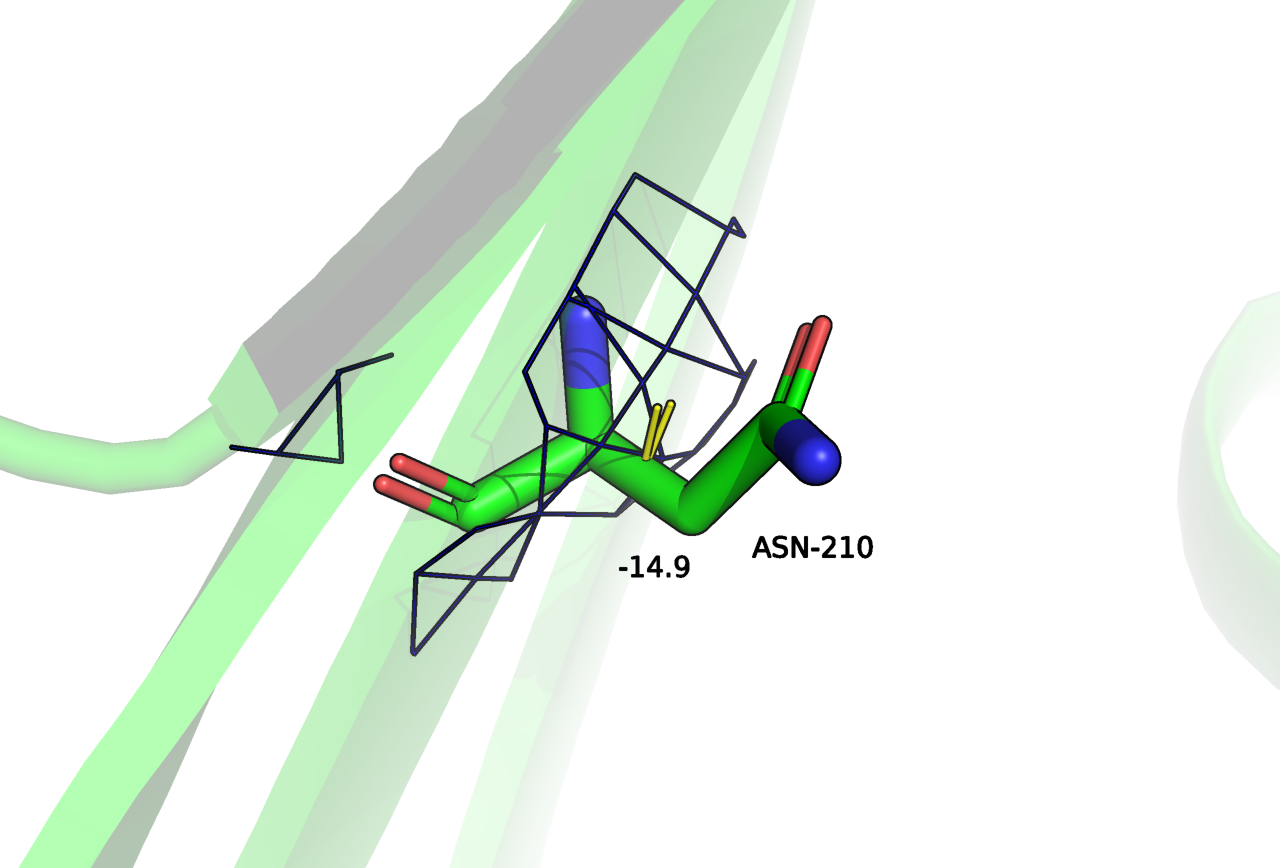
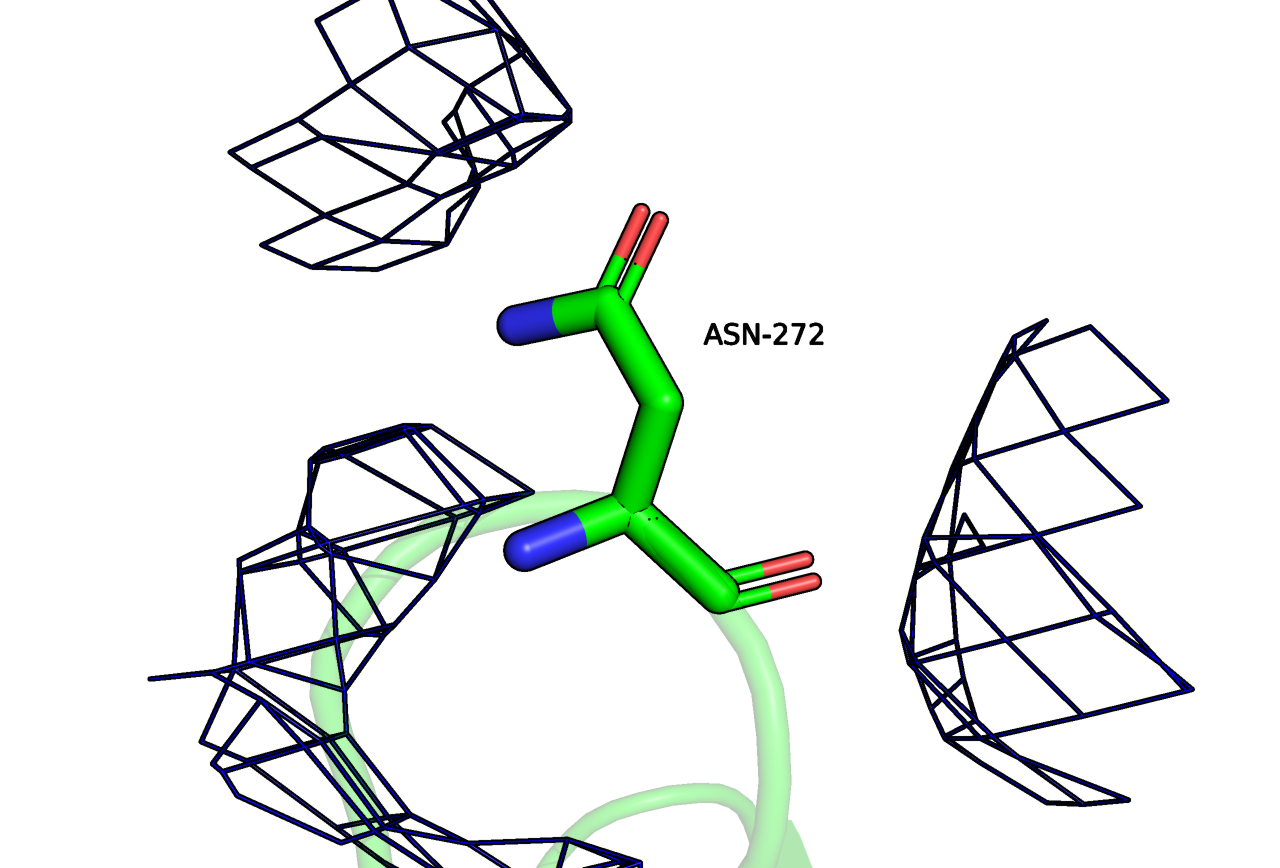
На странице белка в Uniprot можно сделать вывод о функциональной роли отдельных участков. Полезные разделы для поиска позиций: Mutagenesis, Amino acid modifications, Post-translational modification. Я построила mesh вокруг остатков, упомянутых в разделе мутагенез как важные. Важные остатки и mesh вокруг них отражены на Рисунке 5. В целом видно, что поверхности уровня неаккуратные, рваные. Для части атомов вообще участков ЭП нет: у THR-110 некуда вписать боковой радикал, у PHE-193 нет ЭП для кольца, позицию азота остова GLY-274 нельзя определить. Таким образом, такое разрешение позволяет наблюдать общую архитектуру, но к отдельным остаткам много вопросов.
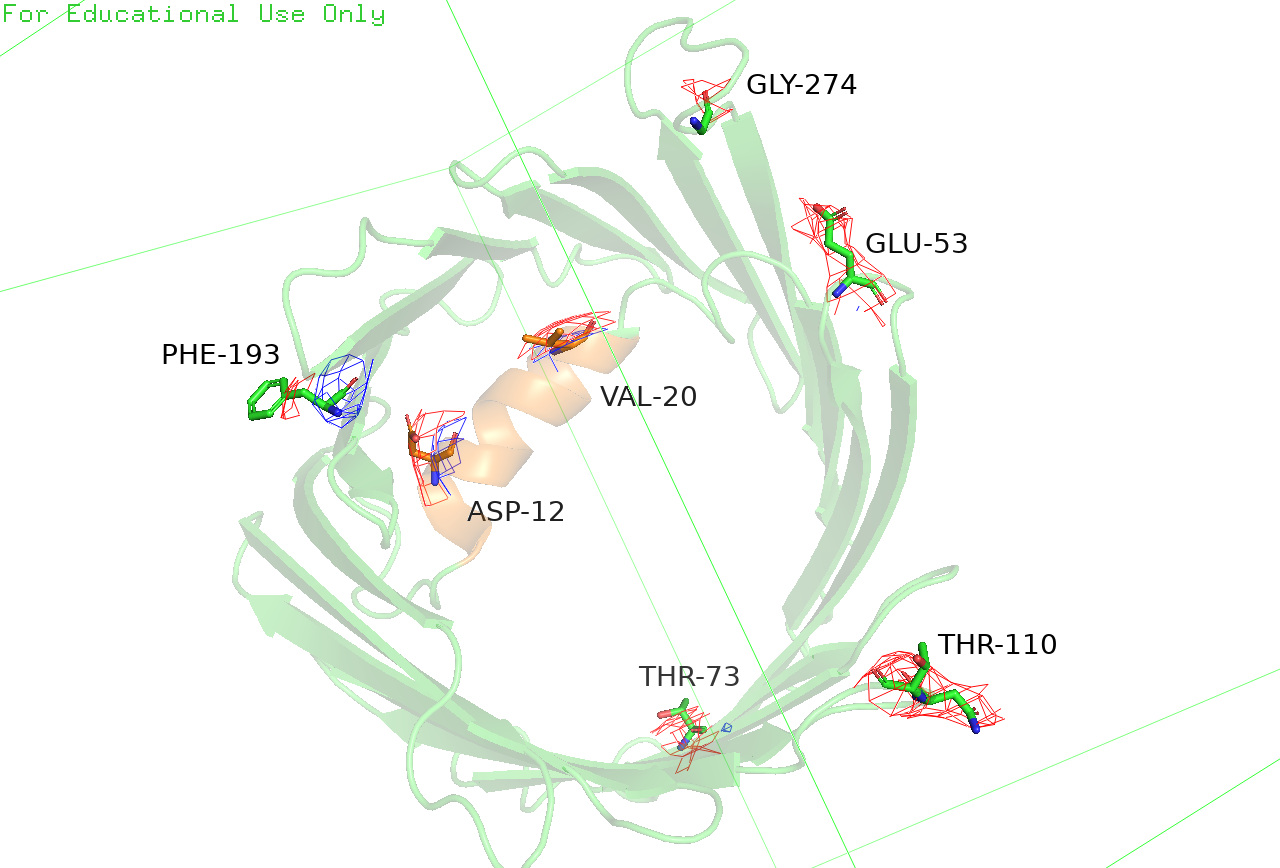
В этом задании я рассматривала остатки-маргиналы по различным характеристикам. Для нахождения маргиналов я пользовалась MolProbity и отчетом PDB. Стоит сказать, что маргиналов очень много и попадаются маргиналы сразу по нескольким критериям.
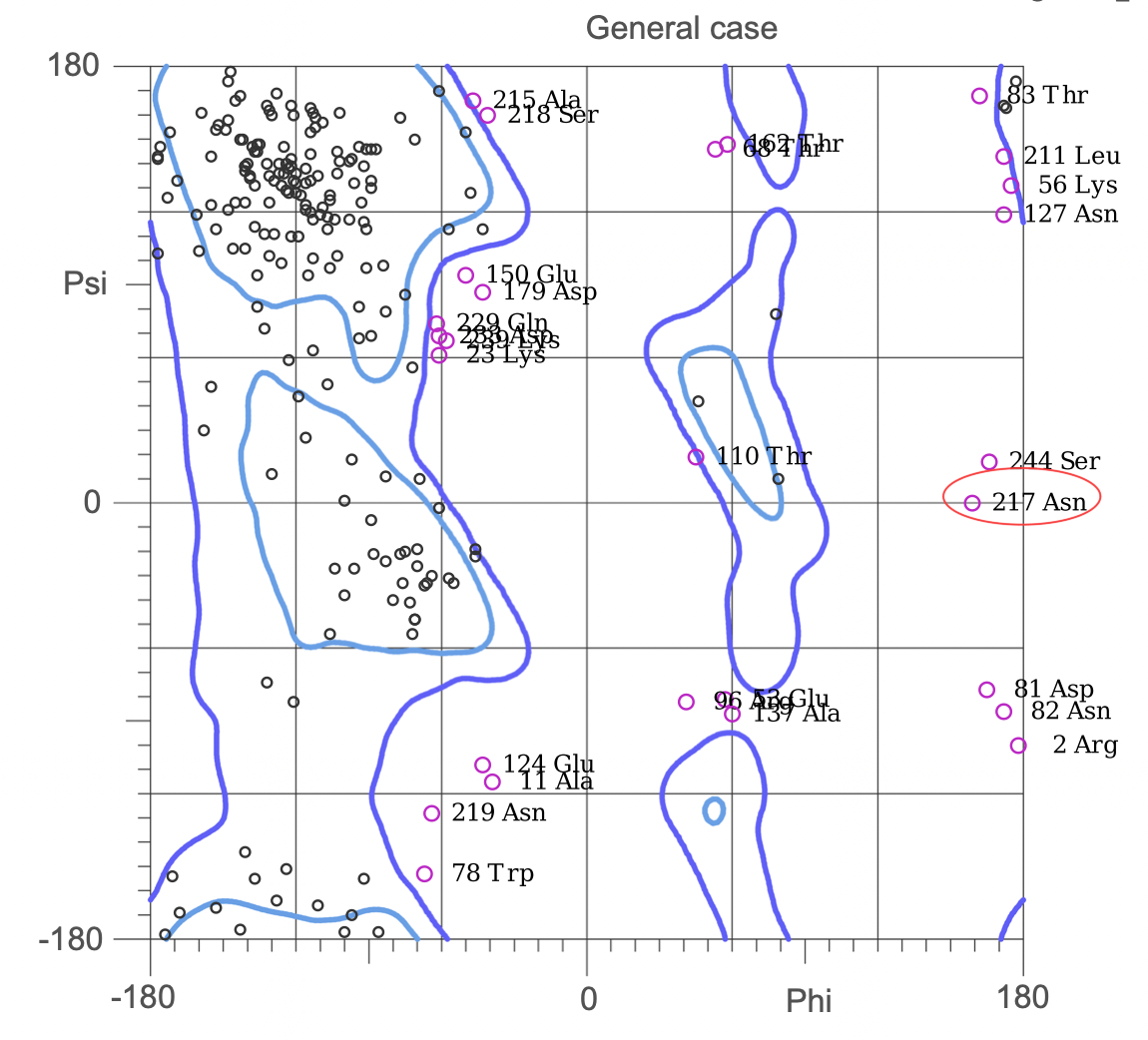
Могу предположить, что данная модель подходит для общего описания архитектуры: анализа числа и расположения консервативных вторичных структур. На отдельные остатки точно полагаться не стоит, в очень многих участков структура не подтверждена ЭП, маргиналы не оправданы. Даже в петле, которая более плотно покрыта ЭП (Рисунок 1) есть маргиналы по нескольким параметрам. В целом всё плохо как для внешних петель, так и для остатков, входящих в состав вторичных структур. Поэтому авторы публикации провели анализ X-ray + NMR для уточнения структуры канала.
В этом задании была предпринята попытка улучшить структуру 2JK4 с помощью сервиса PDB-redo .
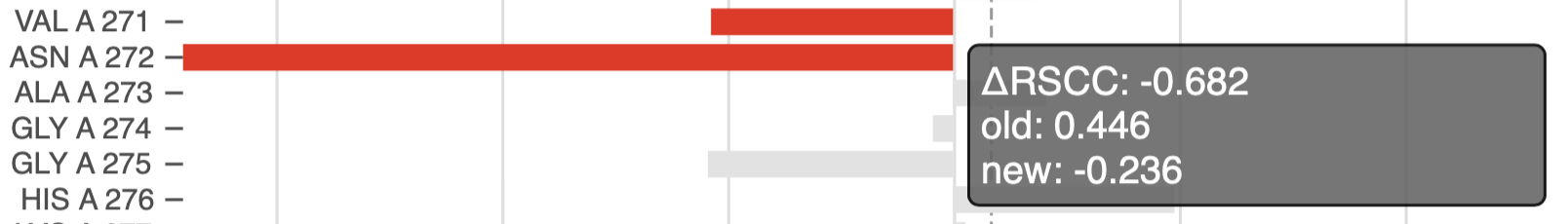
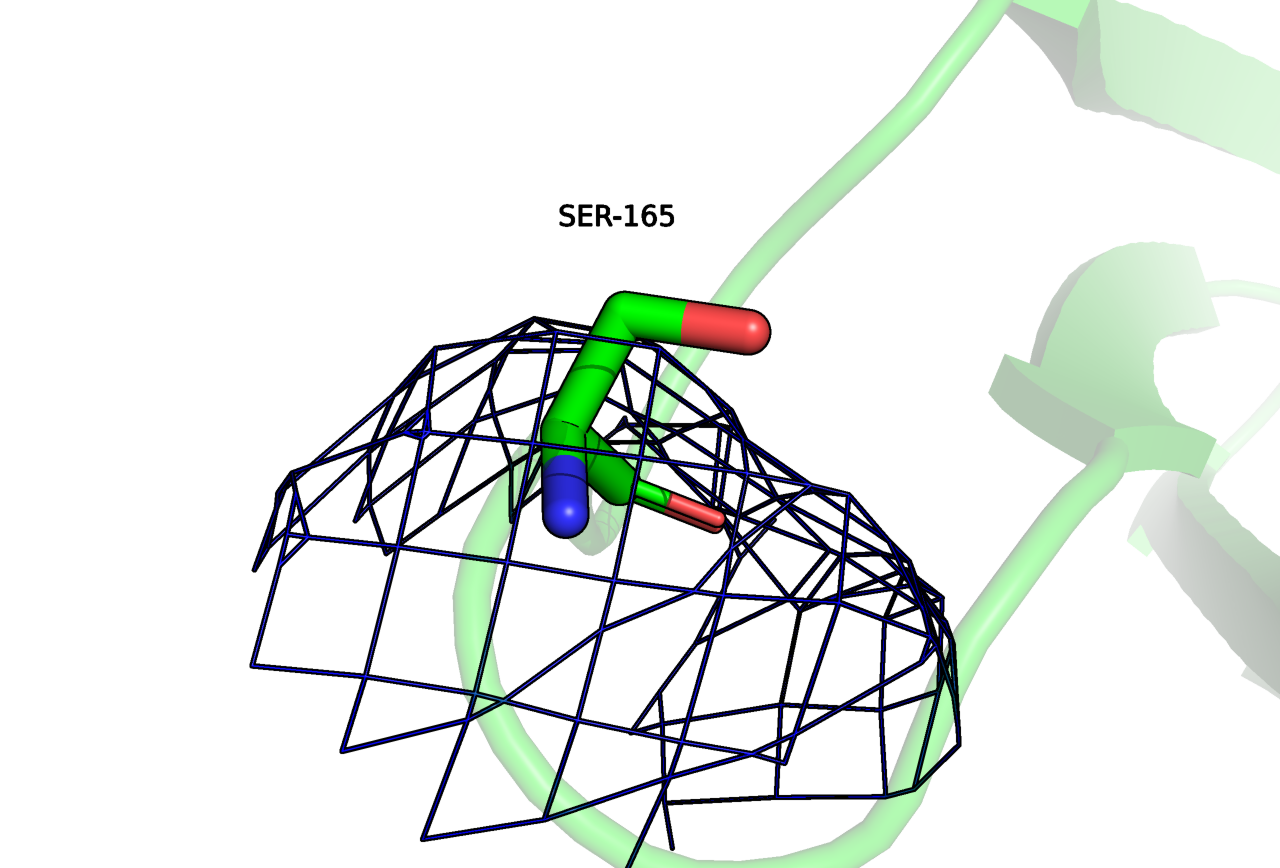
В отчете PDB redo указано, что значение R поменялось с 0.3981 до 0.3695, а Rfree с 0.4777 до 0.4334. Эти значения отличаются от оценок на странице PDB, но независимо от способа подсчета, R-метрики улучшились. В результате отладки структуры расположение 45 остатков стало значимо лучше соответствовать ЭП, но также хуже стали располагаться 6 остатков. На Рисунках 12-13 приведены примеры наиболее значительных изменений пространственного R-фактора в худшую и лучшую стороны в результате преобразований PDB redo. Что "улучшения", что "ухудшения" смысла не несут. Видно, что в первом случае ЭП для остатка нет, во втором случае отсутствует купол для кислорода бокового радикала SER-165.
Интересно, что сильно изменился параметр Bond length RMS Z-score (с 0.171 до 0.543, что существенно) и Bond angle RMS Z-score с 0.477 до 0.735. В идеале эти значения должны быть близки к 1.[5] В отчете PDB не было сообщений о маргиналах по длинам связей, но, видимо, в результате работы программы значения Z-score для связей и углов стали меньше по модулю, то есть меньше отклоняться от ожидаемых средних.
Значительно улучшилось положение остатков на карте Рамачандрана. 22 остатка перешли в предпочтительные регионы по углам (всего стало 237) за счет этого стало меньше остатов в разрешенной зоне (32 против 36) и в запрещенной зоне (17 против 25).
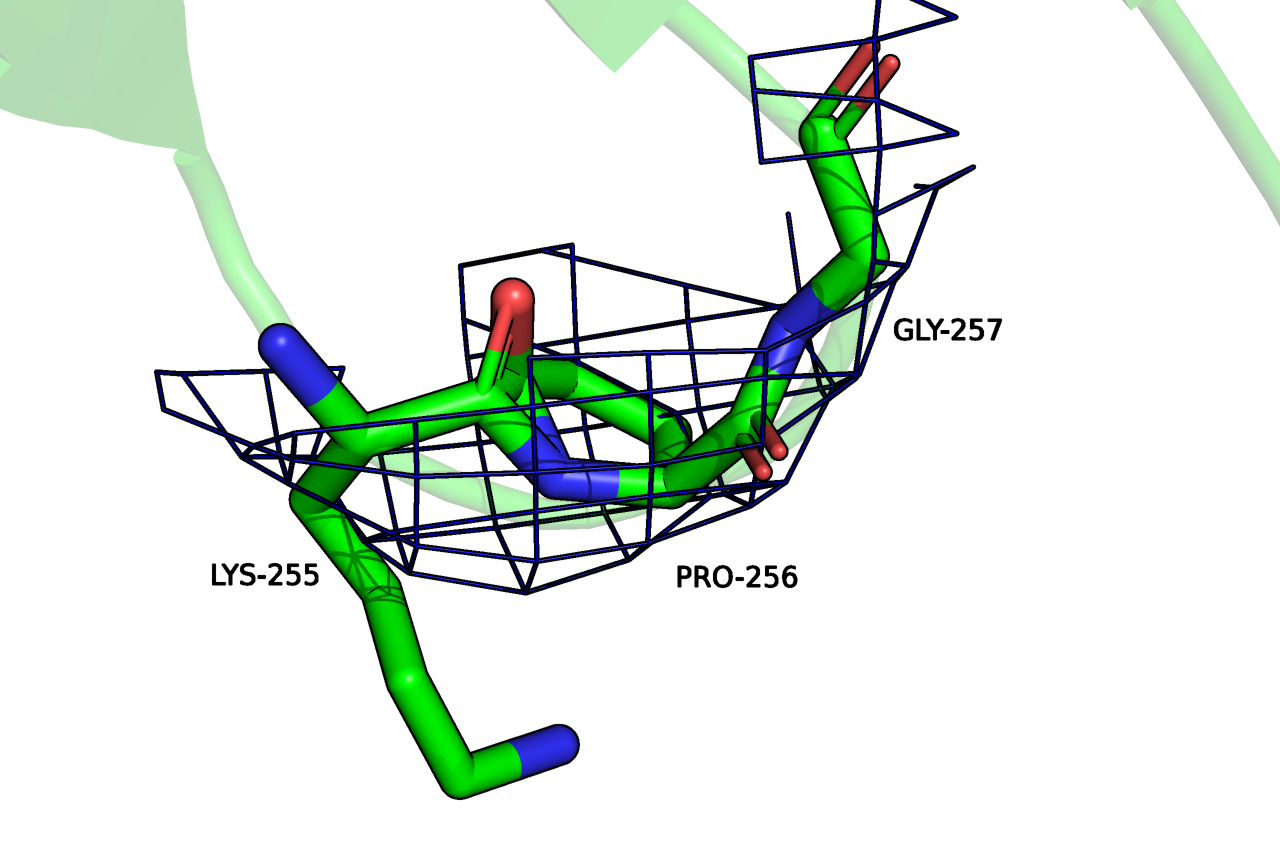
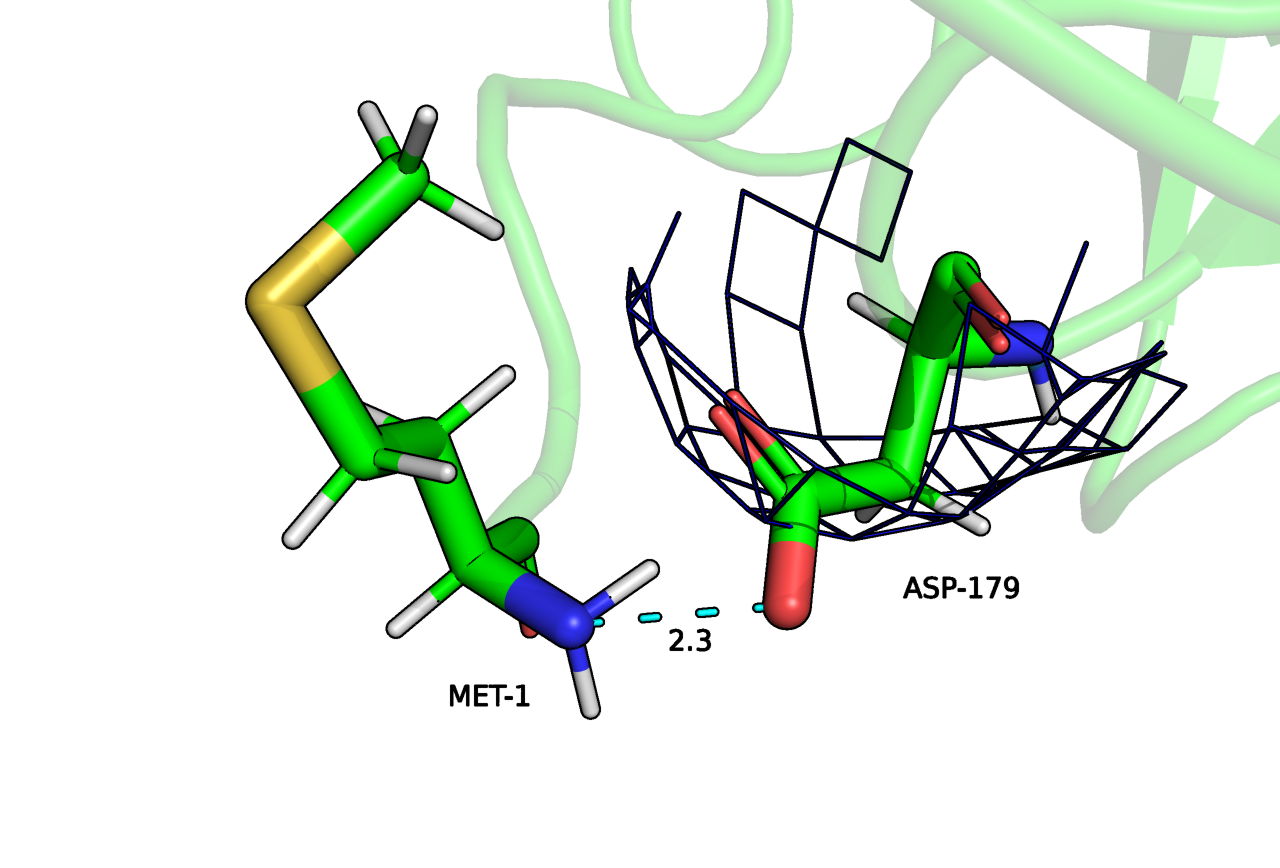
Что касается LYS-255 и PRO-256 с Рисунка 6, по метрикам они стали хуже соответствовать ЭП, это могло произойти в результате "исправления" неверных углов. При этом странный вид в виде лишних связей сохранился, полагаю, что сервис не занимется созданием/разрывом ковалентных связей. По метрикам удалось лучше вписать ASP-179 с Рисунка 7, но clash при этом не был исправлен.
При этом я бы не доверяла улучшениям, внесенным PDB redo, поскольку изначальные данные были плохого качества. Поэтому изменения в метриках здесь часто лишь формальные и от этого структура не становится качественнее. Часто внесенные изменения представляют собой попытки лучше вписать аминокислотные остатки в отсутствие/остатки ЭП. Это еще раз показывает, что на структуру и её части надо внимательно анализировать прежде, чем с ней работать.
[0] The 3D structures of VDAC represent a native conformation
[1]Structure of the human voltage-dependent anion channel
[2] On the complementarity of X-ray and NMR data
[4]The 3D structures of VDAC represent a native conformation
[5] RMS-Z