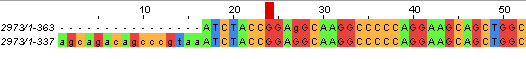
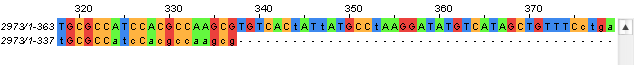
Чтение последовательностей по СэнгеруТекстовое описание:
1.Даны две последовательности: прямая и обратная. Открываем их с помощью двух сопоставленных одна под другой копий программы Chromas Lite. Выравниваем масштабы просмотра с помощью ползунков сбоку и сверху. Обратную последовательность с помощью Reverse+complement в меню Chromas'a переводим в перевёрнутую и комплементарную, чтобы сравнивать с прямой. Ищем в прямой последовательности место, начиная с которого качество чтения становится уверенно выше 10. В последовательности это позиция 28, G. Ищем find'ом строку из следующих 10-12 нуклеотидов (с 28-ого по 40-ой) в обратной последовательности - находим её начиная от 60 нуклеотида.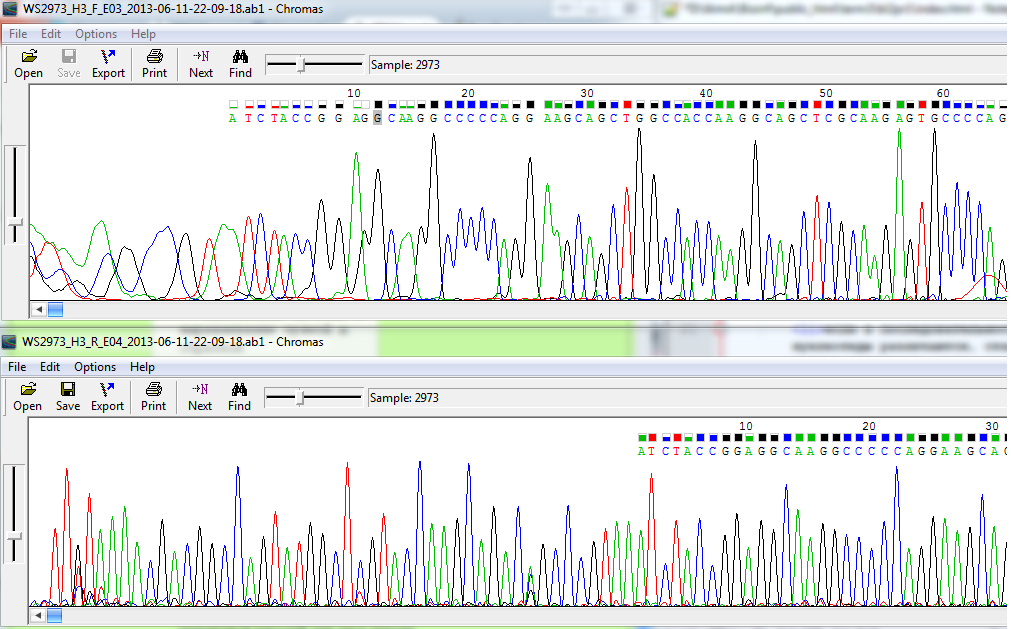 Смотрим на то, что перед 60-ым: прочитано с хорошим качеством, можно попытаться подтвердить то, что прочиталось с плохим качеством в прямой последовательности. 11 нуклеотидов до 28ого в прямой совпадают с предшествующими 60-му в обратной, значит, были определены верно, несмотря на качество прочтения ниже 10. Основания с 0 по 16 в прямой последовательности и с 0 по 48 в обратной (нумерация - по перевёрнутой комплементарной) придётся отбросить.
Теперь приступаем к поиску сомнительных нуклеотидов и доопределению их, исходя из следующих соображений:
Например, в 11ой F (далее прямая последовательность обозначается F, обратная - R) позиции прямой нуклеотид не определён. В R стоит G с качеством 53. Заменяем N в F на G. В позициях 213 и 214 R плохое соотношение сигнал/шум, однако в F нуклеотиды определены с хорошим качеством. Далее в R с 247 по 257-ую позиции наблюдается "пятно краски", F, опять же, прочтена уверенно. По ней, сверяясь с пиками хроматограммы R, восстанавливаем неопределённые нуклеотиды и вставляем в разрыв на 256-ую позицию нуклеотид G. 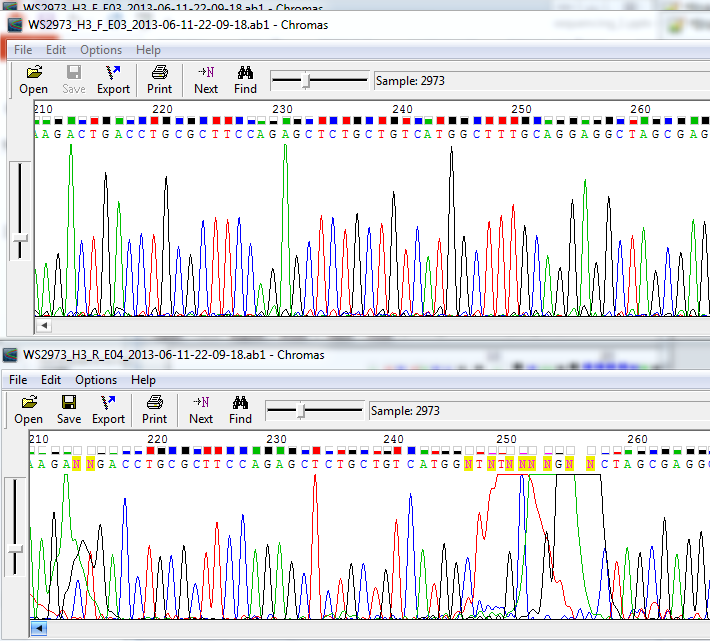 После 276ого качество прочтения в R начинает падать, но остаётся приемлемым для прочтения до 308-ого нуклеотида. Далее до 321 позиции можно идентифицировать нуклеотид, масштабируя уровень хроматограммы и удаляя нуклеотиды там, где их нет по сигналу, а тем, где они есть, но не определены, комбинировать данные сигнала и F. После 321ой позиции R становится нечитаемой: ширины нуклеотидов начинают колебаться в значительных пределах, а пики сигнала выполаживаются, мешая интерпретации данных даже при наличии F для сравнения. Восстанавливаем начало, отброшенное для удобства редактирования, в R вручную (качество сигнала там хорошее). Сверяемся с исходной последовательностью (до удаления её начала при сопоставлении с F): 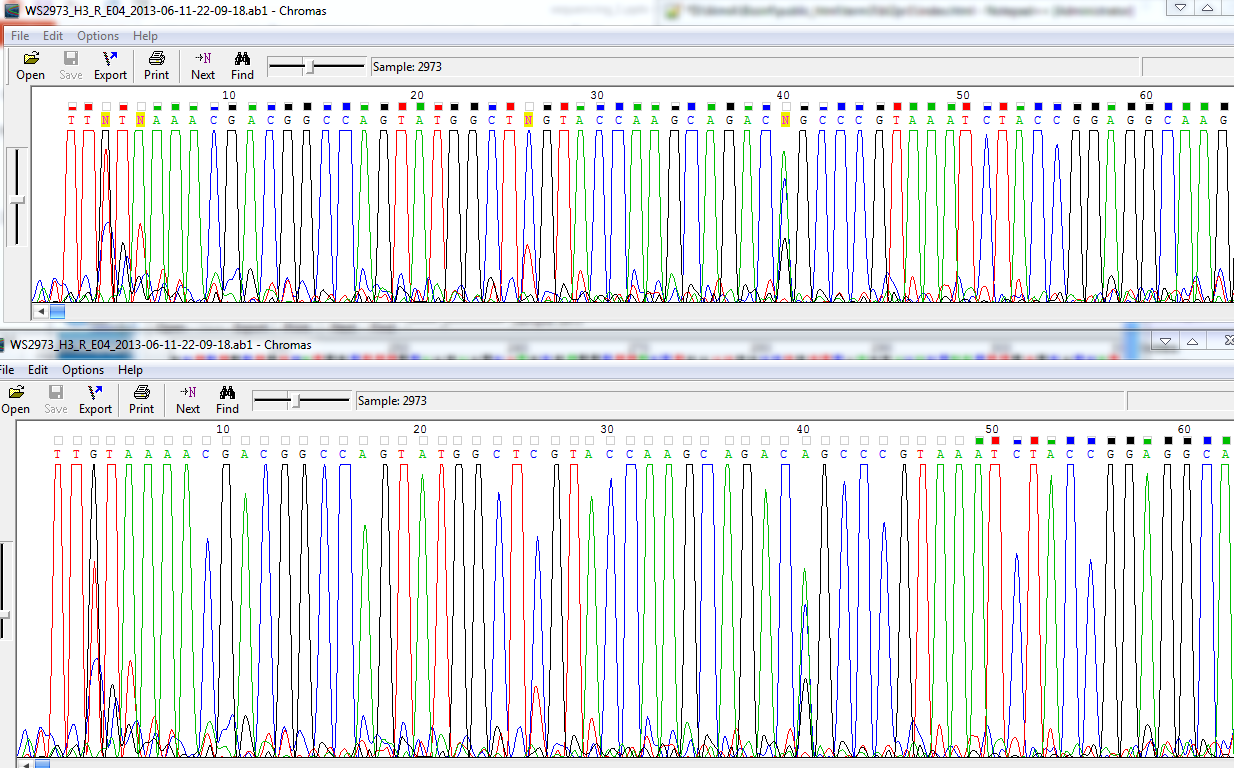 Итоговая длина обратной последовательности составила 369 нуклеотидов.
Итоговая длина обратной последовательности составила 369 нуклеотидов. F интерпретируется по сигналу довольно уверенно вплоть до 363-ей позиции, где по зелёному пику был добавлен ещё один нуклеотид A в конце. Итоговая длина прямой последовательности - 363 нуклеотида. 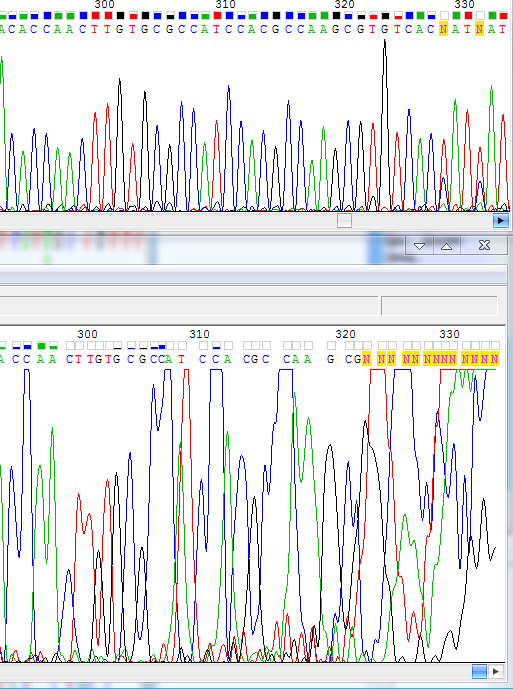
Выравнивание в JalView вручную:
2.Для примера нечитаемой хроматограммы взят этот файл. Начало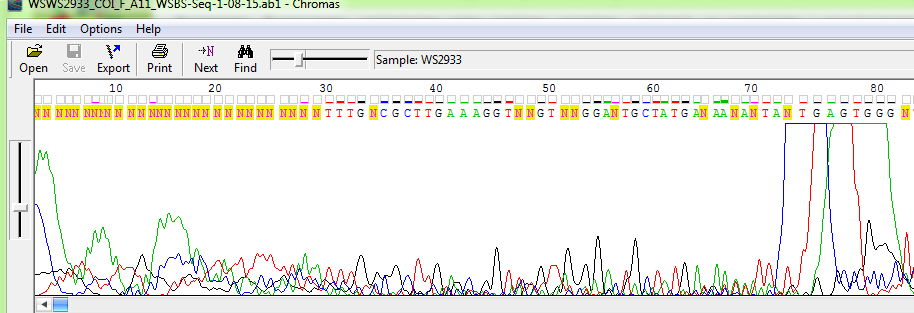 и конец хроматограммы не читаются из-за выположенных пиков сигнала хроматографа, выделить из них соответствие нуклеотиду невозможно. Середина хроматограммы, хотя и содержит чёткие пики, имеет множество посторонних сигналов от примесей (линии ниже основного сигнала), в результате вероятность ошибки в интерпретации сильно возрастает. Возможная причина - плохая подготовка препарата ДНК. и конец хроматограммы не читаются из-за выположенных пиков сигнала хроматографа, выделить из них соответствие нуклеотиду невозможно. Середина хроматограммы, хотя и содержит чёткие пики, имеет множество посторонних сигналов от примесей (линии ниже основного сигнала), в результате вероятность ошибки в интерпретации сильно возрастает. Возможная причина - плохая подготовка препарата ДНК.
|