Сравнение геномов1. Карта сходства двух родственных бактерий
Для сравнения были выбраны Грам-положительные анаэробные мезофильные неподвижные бактерии рода Bifidobacterium (Bacteria; Actinobacteria; Actinobacteria; Bifidobacteriales; Bifidobacteriaceae; Bifidobacterium):
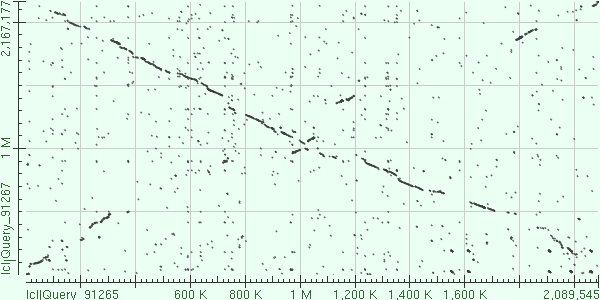 Характеристики выравнивания:
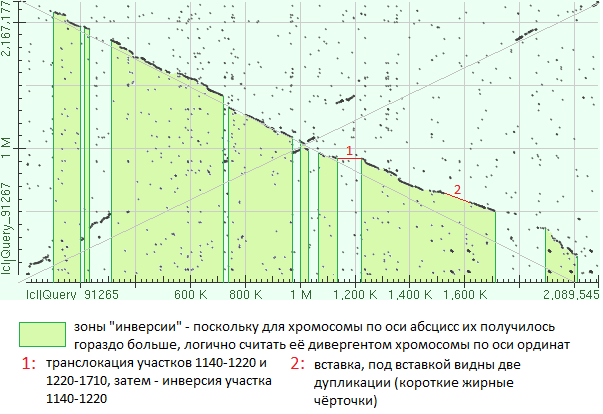
Характеристики выравнивания: 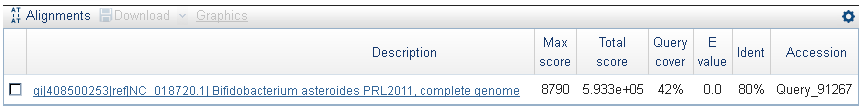 На карте локального сходства можно увидеть крупные инверсии и довольно сложные комбинации инверсий с делециями. Выделила два случая, наиболее крупных (насколько позволил масштаб):
На карте локального сходства можно увидеть крупные инверсии и довольно сложные комбинации инверсий с делециями. Выделила два случая, наиболее крупных (насколько позволил масштаб):
1. Построение пангеномаЗадача, съевшая большую часть времени - нахождение подходящего организма для составления пангенома. Требования: 4 штаммма одного вида, процент идентичности не менее 90, геном каждого штамма секвенирован на уровне не ниже хромосомы. Решение задачи (после всех поломанных копий) выглядит так:
Взяты штаммы FAM18, MC58, Z2491 и 8013. Blastn по 8013 выдал 91% идентичность позиций с остальными штаммами, используем это в качестве настройки. Создаём директорию "Neisseria" в рабочей директории задания, в ней делаем файлы genomes.tsv (сами с помощью любого текстового редактора) и npge.conf (командой npge -g npge.conf), последний корректируем в соответствии с MIN_IDENTITY = Decimal('0.91') и WORKERS = 1 (дабы не грузить сервер). npge Prepare создаст нужные для своей работы файлы и катаогиngpe Examine проверит всё ли в порядке и готово для работы, свяжется с базой и заберёт из неё указанные в genomes.tsv файлы, найдя их по идентификатору. Добавится папочка Examinenpge MakePangenome запустит то, ради чего всё и затевалось - сборк пангенома - в папке Pangenome. Останется дождаться, что случится раньше: сбросится соединение в терминале из-за длитльной неактивности пользователя или всё-же соберётся пангеном Neisseria meningitidis (Пришлось логиниться через tmux: терминал "ушёл" раньше, чем npge завершил работу) npge PostProcessing создаст папки extra-blocks, global-blocks , mutations trees и check с дополнительной информацией о пангеномеСкачиваем результаты работы npge на локальную машину с графической средой, помещаем в папку с ними gnpge.exe и смотрим на получившийся пангеном с помощью gnpge и Excel:
G-blocks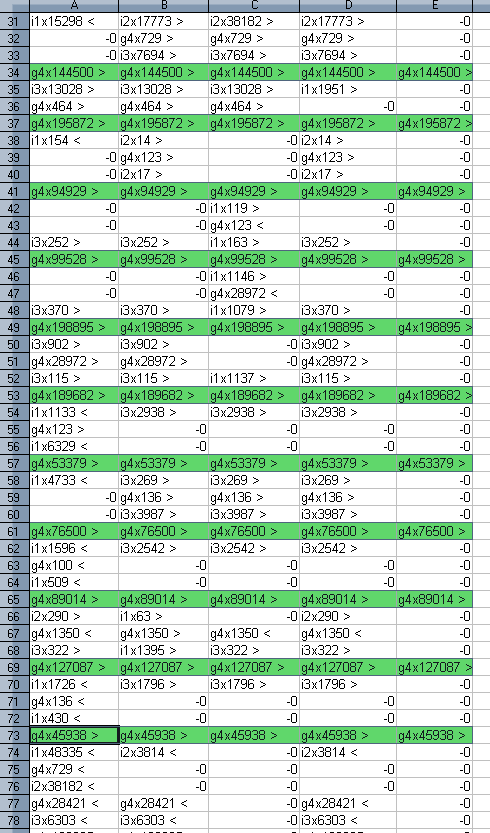 Удалось насчитать 11 консервативных позиций g-блоков, все расположены более-менее кучно в середине хромосом. Также есть перестановки g-блоков: блок g4x28972 у MC58 стоит в позиции 45, а у всех остальных - в позиции 49, или блок g4x123 у 8013 занимает 53-ю позицию, у MC58 - 41ую, у оставшихся - 37ую: 
S-blocksВсего "коровых" блоков (представленных во всех 4х геномах, exact stem blocks, pangenome.info ) 899, по суммарной длине составяющих 61,13% от длины всех блоков и 71,62% от длины генома. Процент консервативных позиций составил 96,3195. R-blocksСамый частый блок - r63x132, встречается 19 рз у 8013, по 13 раз у FAM18 и MC58 и 19 раз у Z2491. Самый крупный блок повторов - r16x5807, у всех штаммов встречащийся по 4 раза. ДелецииСамая крупная по размеру делеция происходит у MC58: выпадает блок h3x3689 (3689 п.н.). Делеция чуть поменьше - h3x2965 - происходит у FAM18. U-blocksБлок u1x132n1 у 8013, расположенный на обратной цепи, хорошо (но не на 100%) выравнивается с трёмя штаммами своего вида, но ни у кого из не аннотирован. Ещё этот блок относительно хорошо выравнивается с FAM18, его прямой цепью, позицями 1160434-1160303: Теперь смотрим в qnpge рядом (картинка выше) - на прямой цепи FAM18 как раз по этим позициям расположился уникальный блок u1x132n2. Ищем гомологов blastn'ом, находим: И, что вполне ожидаемо: Вывод: оба блока вовсе не уникальны, а тот, что в 8013, скорее всего - инверсия с несколькими заменами того, что в FAM18. Аннотация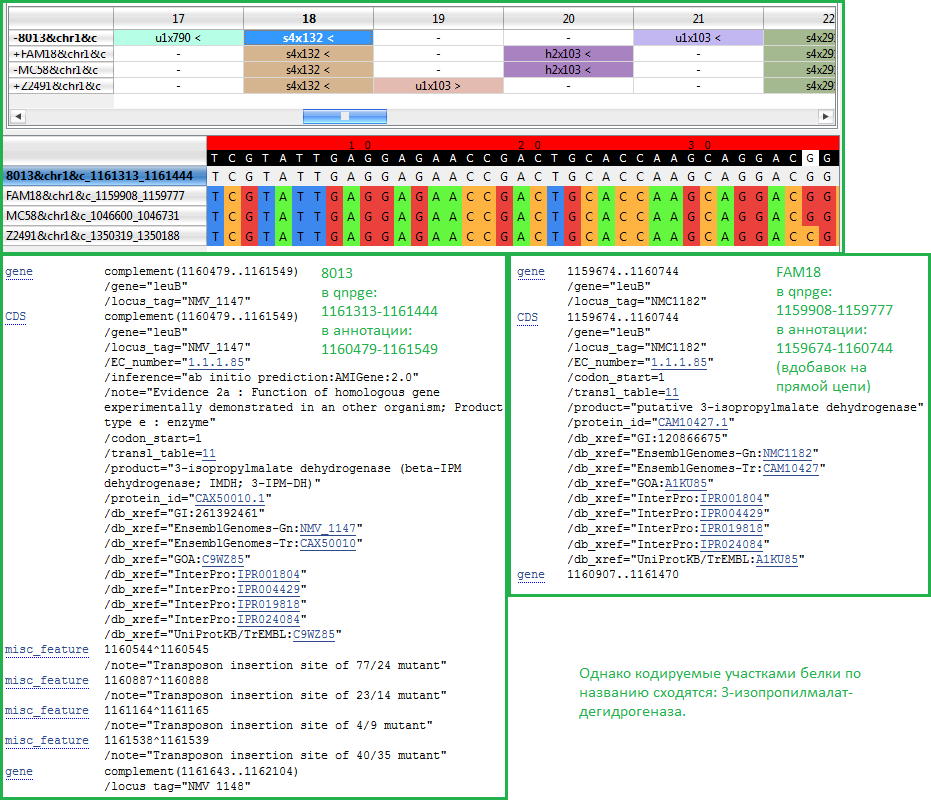 Итоги:Странности результатов npge:  А также невозможность сортировки блоков иначе, чем по возрастанию значений в соответствующем столбце (приходится дополнять Экселем) Впечатления: стало ясно, что не существует универсального или хотя бы сколько-нибудь согласованного алгоритма отыскания "координат" гена. Когда одна программа выделяет одни блоки, а другая, по результатам которой проведена аннотация генов - другие, причём (в моём случае так получилось) проще найти различия, чем сходство - становится определённо непонятно, как использовать полученные результаты. |
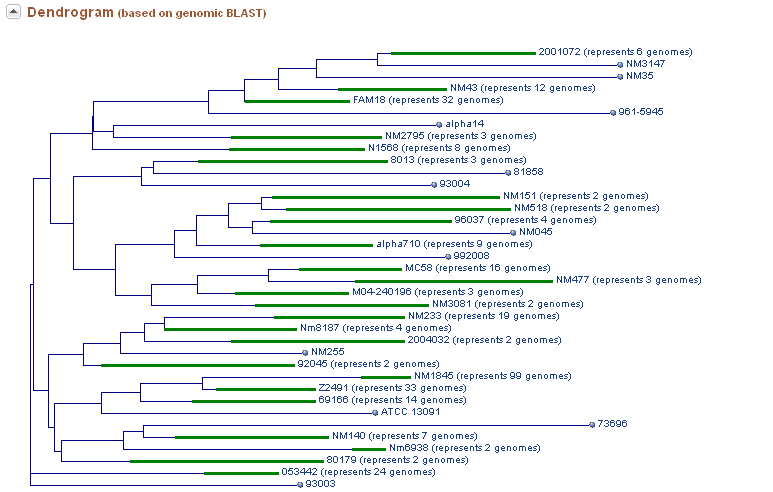
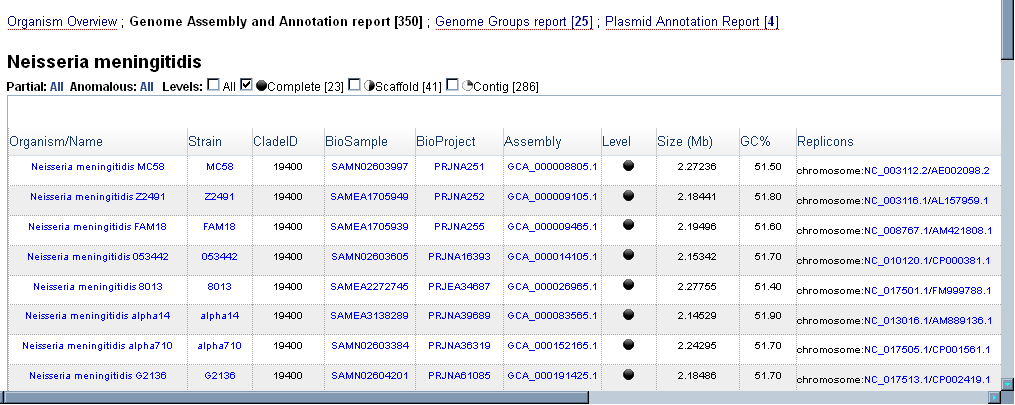 Если меньше 4х сборок, либо все они уровнем ниже - придётся искать ещё. И лучше, наверное, не брать те сборки, которые уже являются пангеномами - они указываются не точками, а чёрточками, длина которых зависит от разброса длин геномов, составляющих пангеном, в разделе dendrogram:
Если меньше 4х сборок, либо все они уровнем ниже - придётся искать ещё. И лучше, наверное, не брать те сборки, которые уже являются пангеномами - они указываются не точками, а чёрточками, длина которых зависит от разброса длин геномов, составляющих пангеном, в разделе dendrogram: