Домены и профили
Построение и проверка работы НММ-профиля семейства белков
Для выполнения практикума было выбрано семейство гликопротеин оболочки ретротранскрибирующихся вирусов HERV-K_env_2 (PF13804). Домен удовлетворяет приведенным в задании критериям: число последовательностей в full состявляет 67, средняя длина домена - 146, покрытие белка доменом оставляет место для второго домена, а также достаточное количество доменных архитектур.
Внутри семейства была выбрана следующая доменная архитектура:

Она удовлетворяет критериям: включает два домена, содержится в более чем 20, но менее чем в половине последовательностей. Такую архитуктуру разделяют 22 последовательности, которые во всех случаях включают 2 домена: HERV-K_env_2 и GP41 (белок оболочки ретровирусов с ID PF00517). Домен HERV-K_env_2 расположен первым. Число белков с данным доменом в выборке Seed, Full и Uniprot составляет 11, 67 и 417 соответственно.
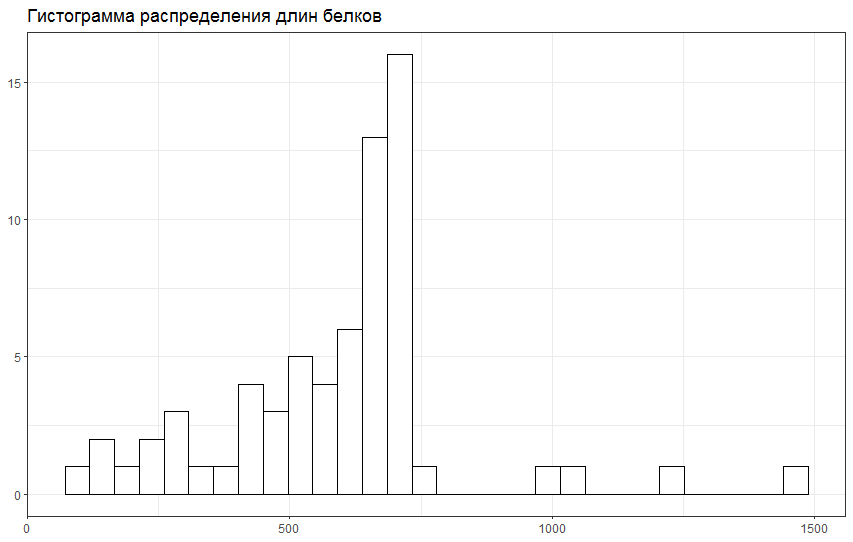
Значительная часть данных и результатов собрана в таблице. Последовательности с выбранной доменной архитектурой были извлечены с помощью скрипта. Ревизованное выравнивание можно посмотреть по ссылке. Было удалено несколько неподходящих последовательностей, а также обрезаны концы. Вид гистограммы, представленной справа, можно объяснить небольшим размером семейства. Наибольшее количество белков находится в диапазоне длины 600-800 аминокислотных остатков, также можно увидеть несколько "аутлаеров" большой длины.
Отредактированное выравнивание передавалось программам пакета HMMER, установленном на kodomo. Для получения данных использовались следующие команды:
Построение профиля: hmm2build hmm_prof.txt edited_aln.fasta
Калибровка: hmm2calibrate hmm_prof.txt
Поиск архитектуры во всех белках, содержащих домен HERV-K_env_2: hmm2search --cpu 2 hmm_prof.txt full.fasta > hmmresult.txt
На выходе был получен файл hmmresult.txt.
HMM-профиль можно увидеть по ссылке. Его длина составляет 634.
С использованием Excel, скрипта на Python, языка R и среды RStudio была создана таблица, приложенная выше. Были расчитаны recall (TPR), precision (PPV) и FPR, а также F1-value.
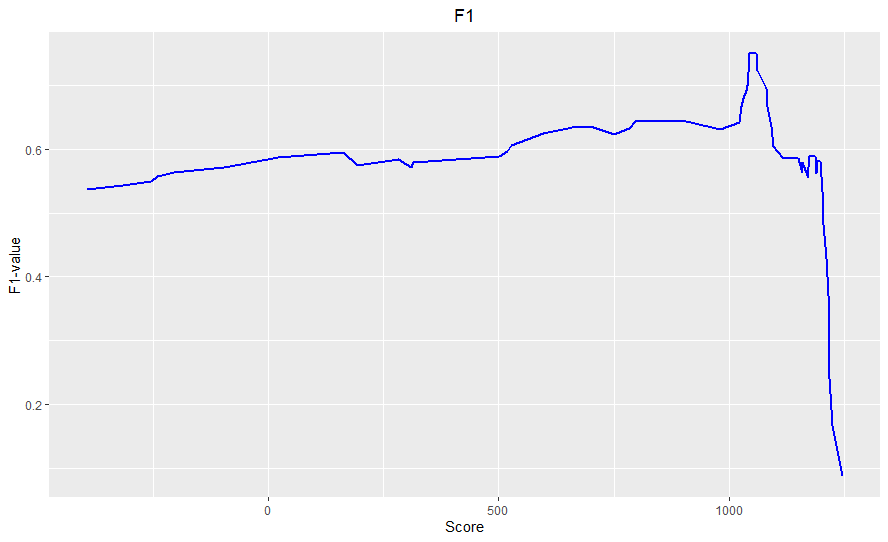
График 1: score для всех белков, отсортированные по убыванию. Эта диаграмма называется Score decrease и показывает, как распределены находки HMM-профиля по их score. Здесь можно увидеть снижение, начиная со значения score 1000.
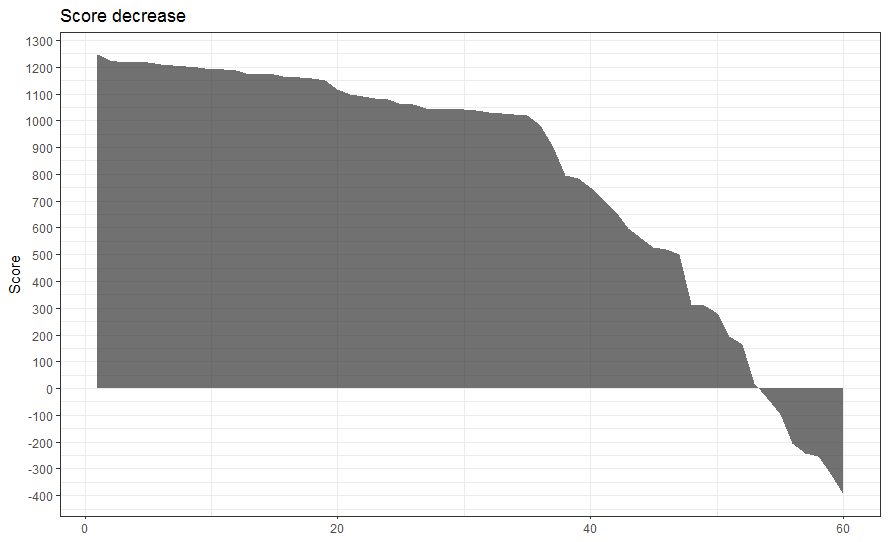
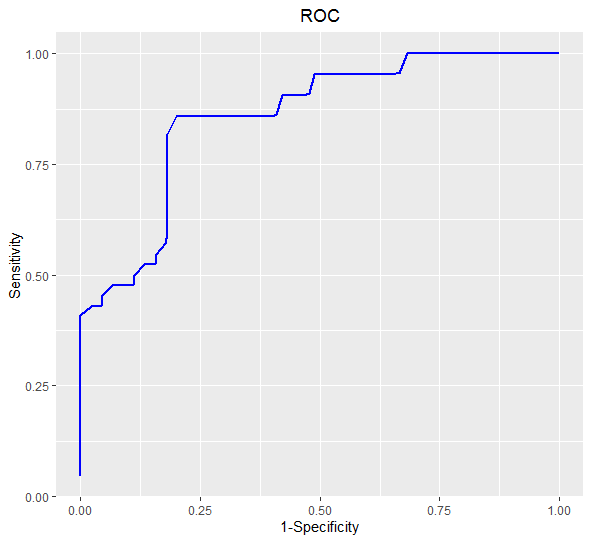
График 2: ROC-кривая, построенная по созданному HMM-профилю. По ней можно найти пороговый параметр, определив точку, соответствующую максимальному расстоянию до диагонали.
Значение функции F1 минимально при нулевой чувствительности, то есть при максимально возможном весе находки, далее резко растет и после достижения пика плавно убывает при уменьшении score к минимальным по выборке значениям. Наибольшему значению F1 0.75 соответствует значение score 1059.7. Данное значение и будет являться порогом.