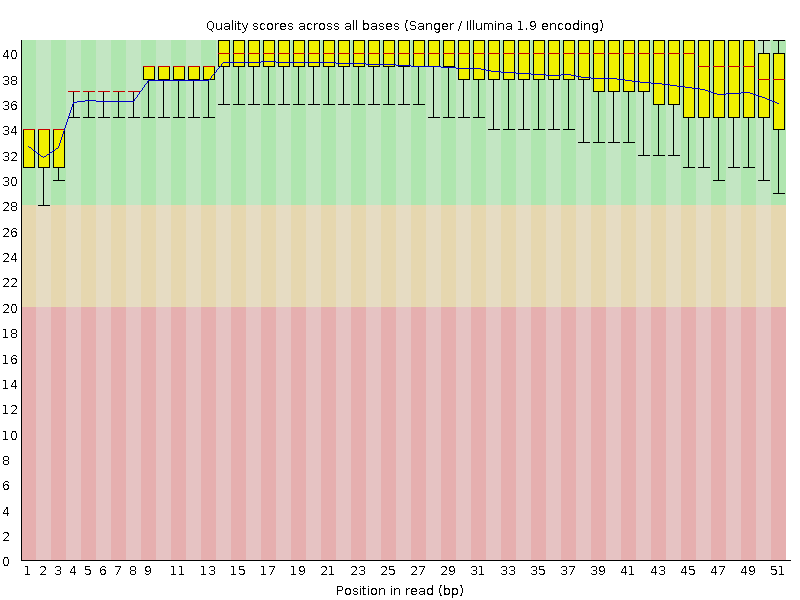
 Рисунок 1. Реплика 1: качество по номеру основания
Рисунок 1. Реплика 1: качество по номеру основания
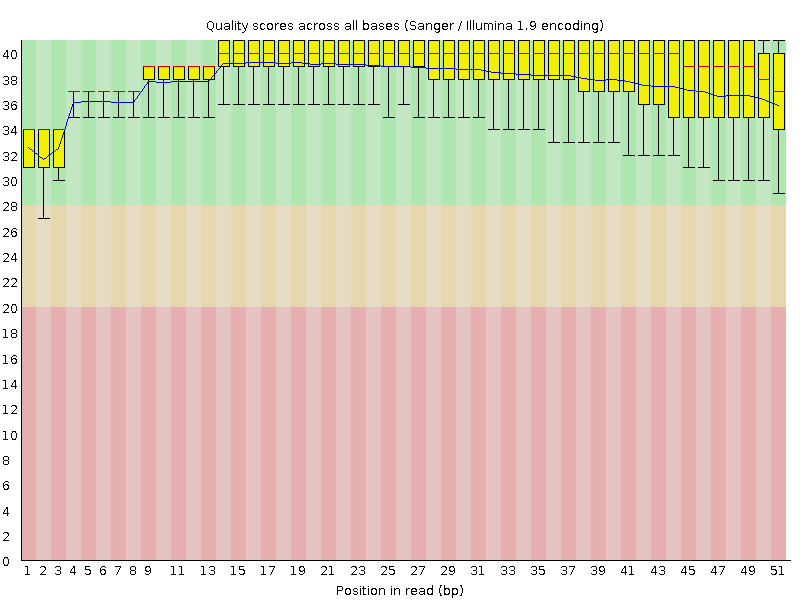 Рисунок 2. Реплика 2: качество по номеру основания
Рисунок 2. Реплика 2: качество по номеру основания
Остальная выдача FastQC имеет незначительные различия; упоминания стоит K-мерное распределение и анализ дубликатов последовательностей:
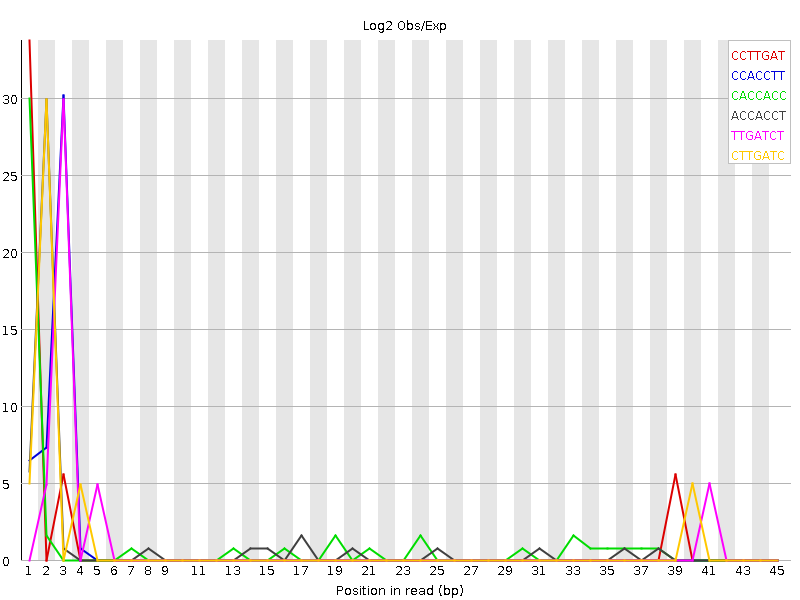 Рисунок 3. Реплика 1: анализ K-меров
Рисунок 3. Реплика 1: анализ K-меров
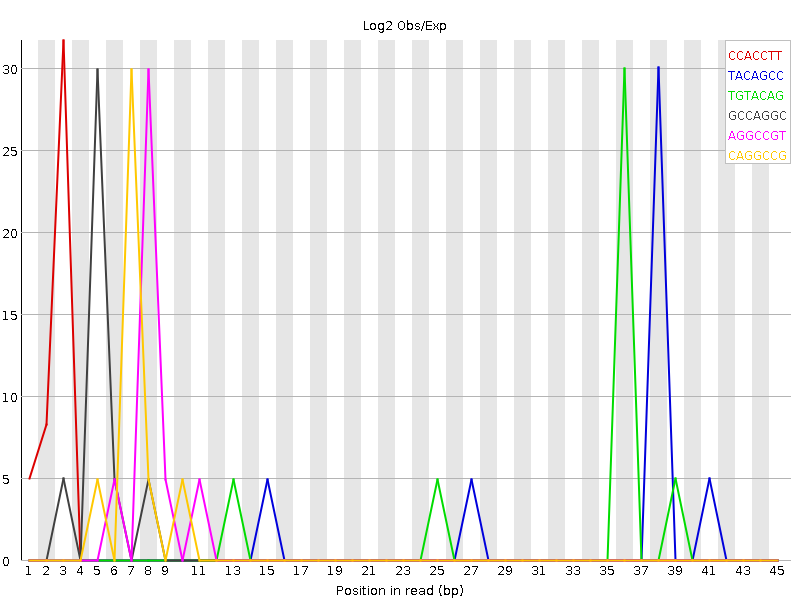 Рисунок 4. Реплика 2: анализ K-меров
Рисунок 4. Реплика 2: анализ K-меров
Из этой выдачи видно, что в репликах встречается несколько отдельных сильно экспрессированных последовательностей – на это указывают отдельные острые пики на графиках.
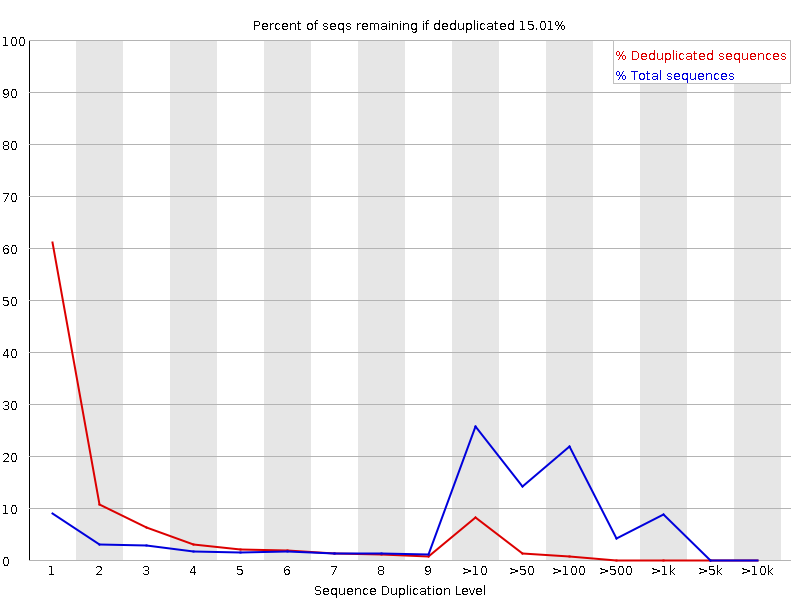 Рисунок 5. Реплика 1: дубликаты последовательностей
Рисунок 5. Реплика 1: дубликаты последовательностей
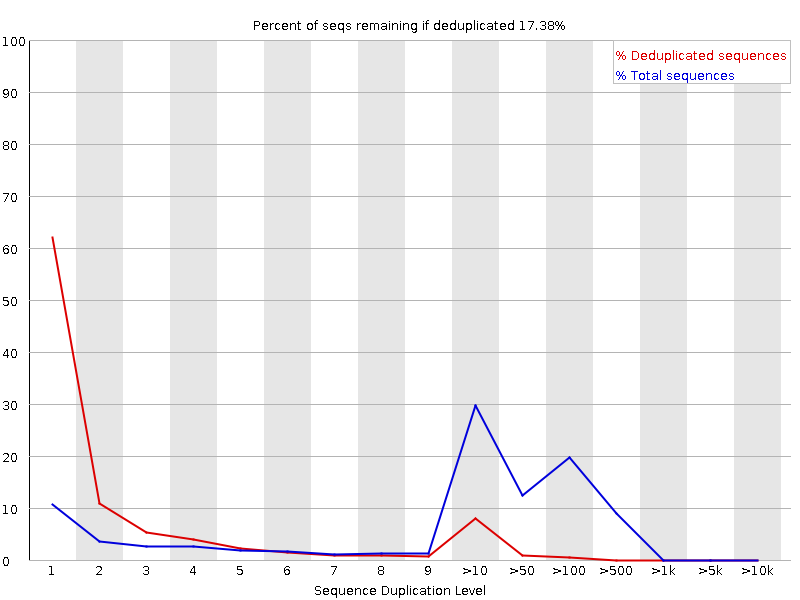 Рисунок 6. Реплика 2: дубликаты последовательностей
Рисунок 6. Реплика 2: дубликаты последовательностей
Видно, что в ридах присутствует очень много повторяющихся последовательностей (особенно в реплике 1) Это весьма характерно для RNA-Seq: для того, чтобы было возможно видеть слабоэкспрессированные последовательности, приходится многократно прочитывать хорошо экспрессированные участки, что создает достаточно большое количество дубликатов чтений.