15. Сборка de novo
Скачивание чтений
wget 'ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/009/SRR4240359/SRR4240359.fastq.gz'
Информация про чтения
Organism: Buchnera aphidicola str. Tuc7 (Acyrthosiphon pisum)
Instrument Mode: Illumina Genome Analyzer II
Reads type: SE (одноконцевые)
Подготовка чтений
1.1 Удаление адаптеров
(прокрутите вправо для просмотра полной программы)
TrimmomaticSE -phred33 ../reads/SRR4240359.fastq.gz output.fq.gz ILLUMINACLIP:adapters.fasta:2:7:7
Результат:
Input Reads: 13557938 Surviving: 13502066 (99.59%) Dropped: 55872 (0.41%)
0.41% чтений оказалось остатками адаптеров.
Удаляем нулеотиды с качеством ниже 20 и последовательности длины
меньше 32:
TrimmomaticSE ../reads/SRR4240359.fastq.gz output.fq.gz ILLUMINACLIP:adapters.fasta:2:7:7 TRAILING:20 MINLEN:32
Результат:
Input Reads: 13557938 Surviving: 12184080 (89.87%) Dropped: 1373858 (10.13%)
Было удалено 10.13% ридов. Размер файла уменьшился с 445M до 385M
2.К-меризация ридов
С помощью команды к-меризуем риды:
velveth Assem 31 -short -fastq.gz output.fq.gz
3.Сборка генома
Собираем геном с помощью команды velvetg:
velvetg ./Assem
N50 сборки: 70607
С помощью команды
cut -f6 stats.txt | sort -h | less -S, находим длины трех самых длинных контигов: 71403 (покрытие - 39.411551, ID - 14),
108447 (покрытие - 42.009184, ID - 1), 125674 (покрытие - 44.550949, ID - 11).
Аномально большое покрытие: 1395.000000, 411220.000000 (их длина =1).
Аномально маленькое: много контигов с покрытием от 1 до 5. Аномальные числа покрытий объясняются тем, что
эти контиги имеют маленькую длниу, которая меньше длины заданного k-мера - 31. При этом они не попадают в contigs.fa.
4.Анализ
Разделяем contigs.fa на отдельные fasta-файлы, чтобы получить fasta-файлы контигов:
seqretsplit -filter contigs.fa dir/name.format
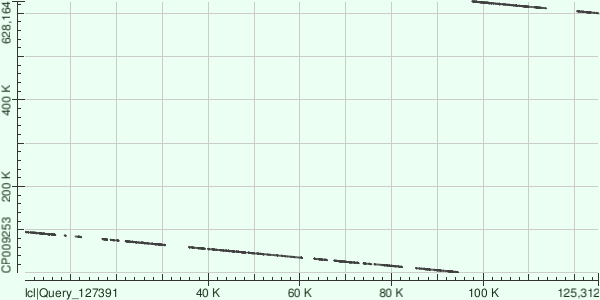
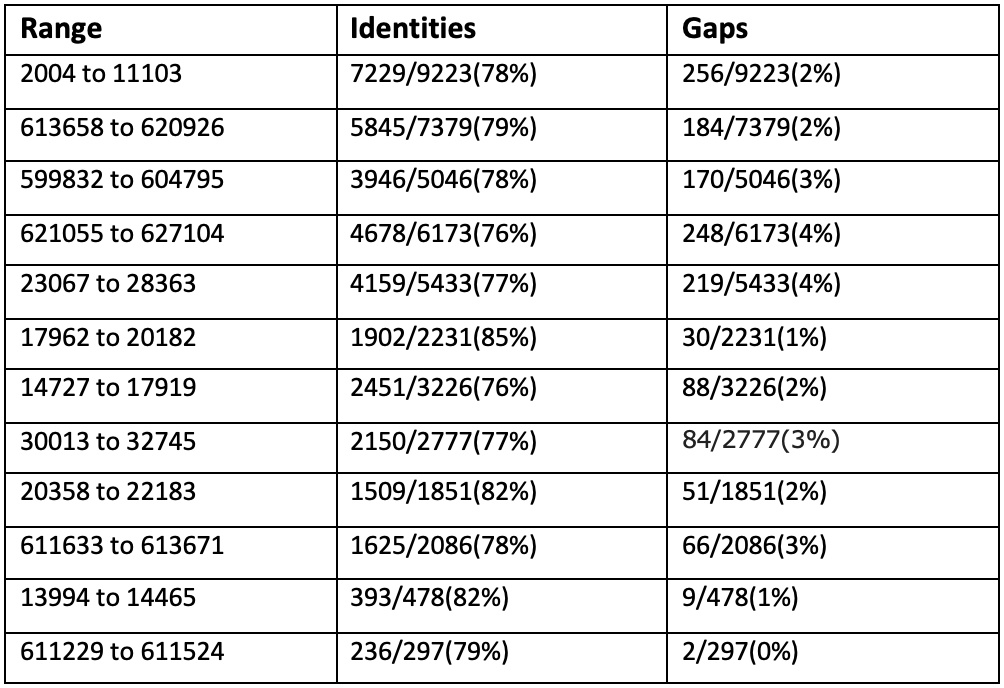
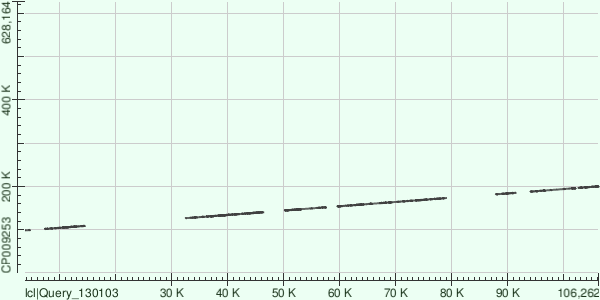
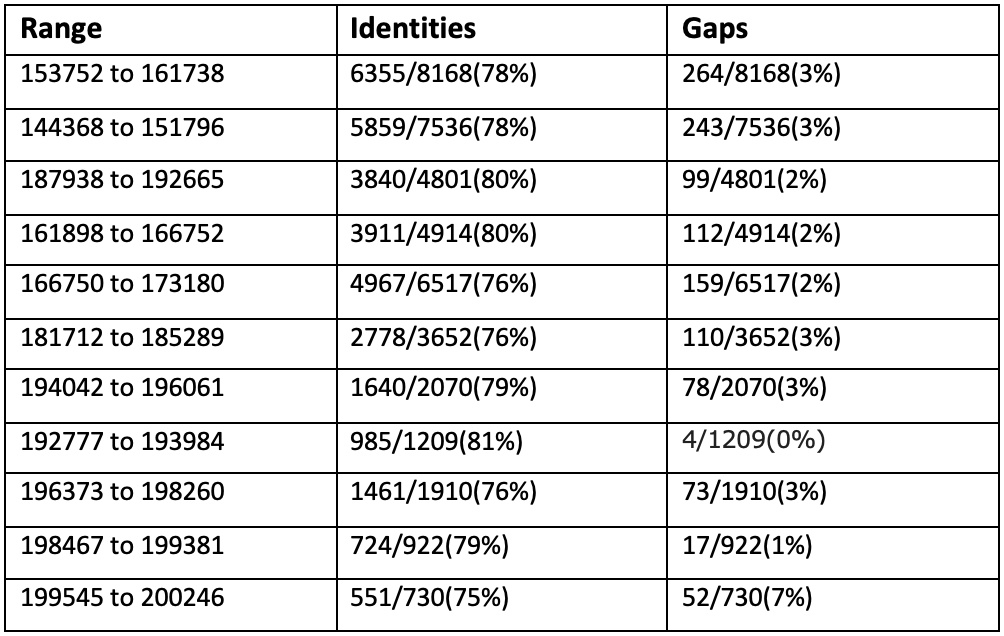
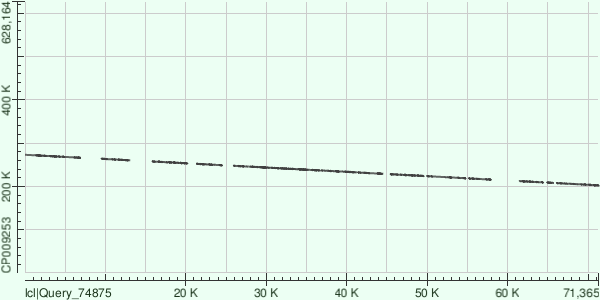
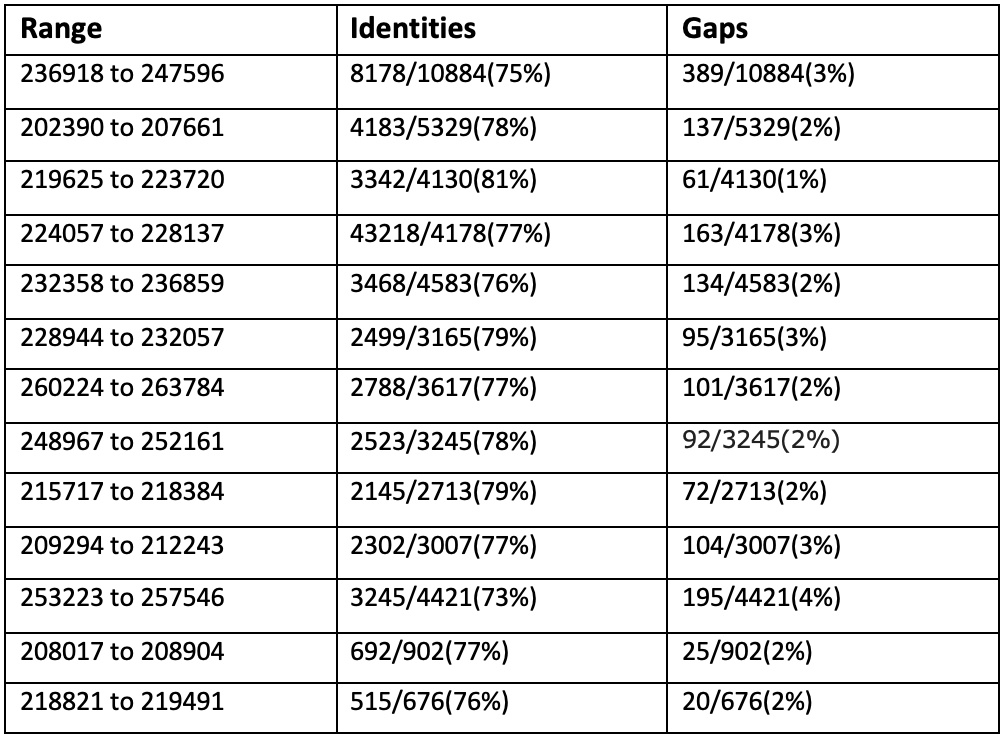
Сравним три самых длинных контига с хромосомой Buchnera aphidicola (GenBank/EMBL AC — CP009253) в megablast: