Для построения HMM-профиля использовались файлы с полными последовательностями белков выравнивания full данного домена, последовательности белков с выбранной архитектурой. Необходимая сортировка по принадлежности белков к архитектуре, а также действия с выдачей hmmsearch проводились в Colab notebook.
Далее с помощью программы множественного выравнивания MUSCLE и Jalview было сделано множественное выравнивание и удаление последовательностей с идентичностью выше 90%. Также были удалены 6 последовательностей с крупными вставками или делециями в доменах архитектуры и выравнивание было сокращено с изначально 1500 символов до фрагмента 770-1350, в котором находятся домены выбранной архитектуры. Файл с результатми можно скачать по ссылке.
Далее я приступил к созданию HMM-профиля.
Список команд для локального запуска программ пакета HMMER на kodomo:
Cоставление HMM-профиля:
hmm2build -g hmmout.txt muscle_edited.faRалибровка:
hmm2calibrate hmmout.txtCравнение белков full с HMM-профилем:
hmm2search --cpu 1 hmmout.txt full_seq.fasta &> hmm_search_result.txt
Файлы с результатами работы программ: hmm_search_result.txt, hmmout.txt. Длина созданного профиля HMM 460 символов. Далее в использованном ранее Colab notebook для каждой находки было определено, принадлежит ли она к выбранной архитектуре, то есть есть ли её AC в списке, взятом со страницы домена Pfam. Результат был экспортирован в Google-таблицы. Там была построена ROC-кривую для результатов работы hmm2search (Рис 2).
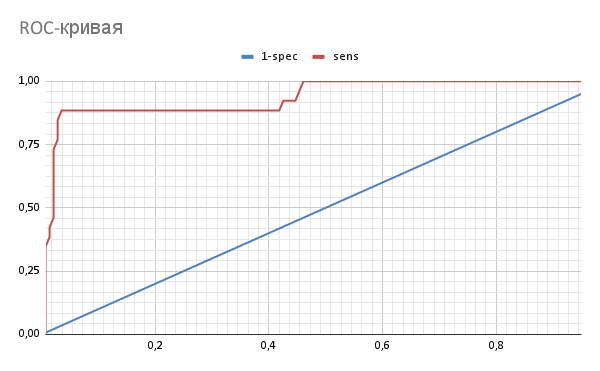
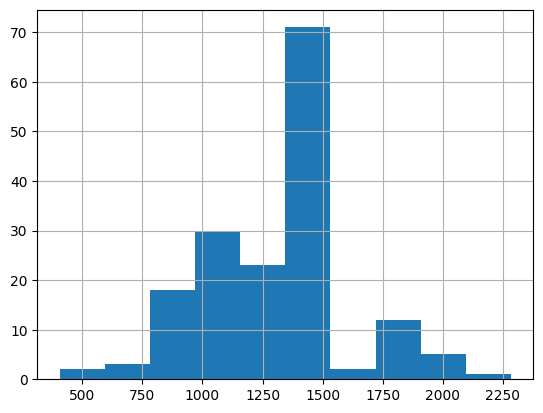
Максимальное расстояние красного графика от прямой y=x находится в точке 1-spec = 0,04, sens = 0,9. Эти параметры могут быть рассмотрены как порог положительного сигнала в находке. Виден резкий скачок в чувствительности, при малом изменении 1-специфичности, по этой причине в качетче порога был выбран параметр sens = 0,9. По нему была построена колонка "Сигнал +". Визуально сигнал почти везде совпал с данными о наличии архитектуры в находке. Значит, выбранный порог оказался удачным. Диаграмма распределения длин белков в full представлена на Рис 3.